INTRODUCTION
Chlorine-based chemistry has a double impact on the environment: a direct one due to its toxicity and an indirect effect related to the industrial production of the chlorine molecule. This latter process is highly energetically intensive (electrolysis of sodium chloride), and it releases ca 2.1 tons of CO2 per tons of product. As a comparison, the cement or the steel industries release “only” 0.92 and 1.8 tons CO2/tons product, respectively (Table 1).
| Industrial sector | Tons of CO2/tons of product |
| Cement industry | 0.65–0.92 tons CO2/tons cement |
| Ammonia production | 1.15–1.30 tons CO2/tons ammonia |
| Steel production | 1.8 tons CO2/tons steel |
| Chlorine production | 2.1 tons CO2/tons of chlorine |
In this prospect, the use of safe-to-handle and beyond-chlorine substitutes is highly desirable [4–6]. Dimethyl carbonate (DMC), nowadays produced by a clean and halogen-free process [7–9], is a well-known environmentally benign substitute of phosgene in carboxymethylation reactions (via BAc2 mechanism) and DMS or methyl halides in methylation reactions (via BAl2 mechanism). DMC has also numerous applications as nontoxic solvent and reagent [10–20].
In general DMC, as a methylating agent requires temperatures higher than 150°C in the presence of a base and an appropriate nucleophile [21]. In fact, when hard alkoxides are reacted with DMC at high temperature, they result exclusively in the transesterified methylcarbonates derivatives [22]. On the contrary, soft nucleophiles such as anilines, phenols, and methylene-active compounds are easily methylated by DMC via a BAl2 mechanism [23–26].
Methyl ethers of primary alcohols can be obtained through a two-step reaction: a BAc2 transesterification followed by decarboxylation of the resulting methylcarbonate [12]. An interesting exception is the one-pot synthesis of 5- and 6-membered heterocycles by DMC chemistry comprising of a carboxymethylation reaction (BAc2) followed by an intramolecular alkylation (BAl2) both occurring at reflux temperature [27–30]. In this case, computational investigation has demonstrated that the formation of the cyclic ethers is energetically the most favorable pathway due to a large entropy reduction that occurs at the relevant transition states.
It is also noteworthy that methylation of secondary alcohols is, generally, never obtained quantitatively due to the formation of the predictable elimination products [22].
However, recently, we have reported the reactivity of the secondary hydroxyl groups of bio-based platform isosorbide that can be quantitatively methylated at reflux temperature (90°C) by reaction with DMC in the presence of a stoichiometric excess of a strong base [31].
This reaction is particularly relevant as it encompasses the transformation of a renewable source derivative into an industrially appealing product by beyond-chlorine chemistry.
It is also noteworthy that over the last 10 years anhydro sugar alcohol isosorbide has gained growing interest as bio-based platform chemical as its derivatives have shown potential applications in substituting fossil resource-based products [32–33].
Isosorbide, namely, 1,4:3,6-dianhydro-d-glucitol, can be produced from biomass by a multistep reaction [34–37]: first glucose is obtained by polysaccharides depolymerization, then this sugar is converted into sorbitol by hydrogenation reaction. Finally, isosorbide is synthesized by a twofold dehydration acid-catalyzed reaction of sorbitol via 1,4-sorbitan intermediate (Figure 1 [38–39]). Recently, a milder synthesis of isosorbide starting from D-sorbitol and DMC has also been reported [40].

Isosorbide has a considerable potential for the production of versatile new chemicals as its hydroxyl groups can be easily functionalized or directly processed (Figure 1).
Mono- and dinitrate isosorbide, for instance, have found applications as vasodilating pharmaceuticals for the treatment of angina pectoris [41, 42], isosorbide alkyl esters are used as replacements for plasticizers [43–46], short-chain aliphatic isosorbide ethers are employed as coalescents in the paint industry [47–50], and isosorbide derivates have also been investigated as fuel additives.
Furthermore, aliphatic/aromatic esters and ethers of isosorbide have showed diverse and useful properties as new compounds. Applications of interest include, green solvents with high boiling points (dimethyl isosorbide, DMI [51]) and surfactants (monosubstituted isosorbide).
Most importantly, isosorbide and its derivates are used as monomers in the manufacture of several polymers and copolymers [52]. Few examples are poly-(ethylene-co-isosorbide)terephthalate (PEIT), poly(isosorbide oxalate), and poly(isosorbide carbonate [53–57]).
The interest of the chemical industry in this anhydro sugar is not only due to its bio-based nature but also to its high reactivity and peculiar molecular structure.
Isosorbide has a characteristic V-shaped configuration formed by two cisconnected tetrahydrofuran rings with an opening angle of 120° (Figure 2) where each hydroxyl group is in the β-position to both furanic oxygens. This molecule comprises of two secondary hydroxyl moieties in the 2- and 5-positions, the OH group labeled as endo is directed toward the V-shaped cavity, whereas the exo OH group is pointing outside of the sugar cavity.

The configuration of the two hydroxyl groups is extremely important for the reactivity of isosorbide. In fact, isoidide and isomannide, the two epimers of isosorbide that incorporate only exo or endo hydroxyl groups respectively (Figure 3), show different physical and chemical properties compared to isosorbide and, most importantly, diverse reactivity of the hydroxyl groups [58].

Isomannide and isoidide are generally synthetized by isosorbide isomerization, although several studies are ongoing at the moment so to render this process more economically viable [59, 60].
Isomannide, is not a very attractive reagent as the two endo hydroxyl groups show a low reactivity [53]. Furthermore, its production in large scale is expensive compared to isosorbide since it requires fructose or mannitol as starting material.
Isoidide, the third epimer, cannot be obtained from plant biomass as its main precursor is L-idose.
It should be also mentioned that, despite being bio-based platform molecules, chemical transformations involving isosorbide and its epimers still involve mainly halogen-based harmful chemistry [61–64].
In this context, the remarkable reactivity of isosorbide with DMC, used as beyond-chlorine methylating agent, is quite relevant. According to our previously reported data, the secondary hydroxyl groups of isosorbide were quantitatively methylated at reflux temperature (90°C) by reaction with DMC in the presence of a stoichiometric excess of a strong base [28]. This result was ascribed to isosorbide's unique structure. In fact, within isosorbide “V”-like configuration, each hydroxyl group is in the β-position to both furanic oxygen and this vicinity might enhance its reactivity toward DMC. Furthermore, it has been demonstrated that the nucleophilic character of the hydroxyl oxygen atom in the 5-position (endo) of isosorbide is enhanced as a result of the intramolecular hydrogen bond (Figure 2). Thus, the endo hydroxyl group in 5-position is methylated more easily than the exo in 2-position at reflux temperature. In order to understand the influence of the molecule backbone of secondary alcohols in BAl2 reaction, a number of secondary alcohols of similar structure were selected and their methylation investigated; however, among these compounds, isosorbide showed by far the higher selectivity toward methylation. This result was ascribed to the neighboring effect of the oxygen in β-position and the presence of the intramolecular hydrogen bond at the endo hydroxyl group.
In this prospect, the aim of the present work is dual: the synthesis of isosorbide derivatives with potential industrial applications, i.e. DMI as a green solvent and dicarboxymethyl isosorbide (DC) as a monomer for polymers preparation and then compare, under the best found conditions, the reactivity of the two epimers isomannide and isoidide.
A complete study on carboxymethylation (via BAc2 pathway) and methylation (via BAl2 pathway) reactions via DMC chemistry of isosorbide has been conducted. Reaction conditions have been optimized using either an autoclave or performing the synthesis at reflux temperature. Thus, in order to better understand the nucleophilicity properties of the hydroxyl groups, the reactivity of isosorbide epimers isomannide and isoidide have also been investigated. Interestingly, the results showed an opposite reactivity trend of the three epimers in base-catalyzed methylation compared to carboxymethylation reactions.
RESULTS AND DISCUSSION
Carboxymethylation reaction of isosorbide and its epimers
Carboxymethylation of isosorbide was investigated employing DMC as a reagent and a solvent via a BAc2 mechanism according to Scheme 1. Earlier investigations have showed that potassium carbonate, used in stoichiometric excess (at least in 2 mol. eq.) was the best base for the reaction [27–30].

In our first experiment, isosorbide was dissolved in DMC (30 mol. eq.) in the presence of a stoichiometric amount of K2CO3 (1 mol. eq.), and the mixture was refluxed at 90°C. Consistently with our earlier results, gas chromatography–mass spectrometry (GC-MS) analysis showed that, after 6 hours, isosorbide was completely converted, although the selectivity toward the DC was only moderate (54%). Monocarboxymethyl derivatives MC1 and MC2 were still present in the reaction mixture in 37% and 9%, respectively (Entry 1, Table 2).
| # | K2CO3 (eq. mol.) | Conv. (%) | Selectivity (GC-MS) % | ||
|---|---|---|---|---|---|
| DC | MC1 | MC2 | |||
| 1b | 1.00 | 100 | 54 | 37 | 9 |
| 2 | 1.00 | 100 | 85 | 10 | 5 |
| 3 | 0.50 | 100 | 85 | 11 | 4 |
| 4 | 0.25 | 100 | 87 | 10 | 3 |
| 5 | 0.10 | 100 | 90 | 8 | 2 |
Note: Bold values indicate the wanted product.
Reaction conditions: isosorbide: DMC 1:30 eq. mol.; temperature 90°C; reaction time 6 h. All the reactions have been conducted in anhydrous conditions.
The reaction has not been conducted in anhydrous conditions.
The reaction has then been repeated working in anhydrous conditions, i.e., isosorbide was kept under vacuum overnight, K2CO3 was dried in a oven at 100 °C, and DMC was dehydrated on molecular sieves. Furthermore, all the glassware has been accurately dried with a heatgun, and the reaction was conducted under nitrogen atmosphere. In these reaction conditions, after 6 hours, the selectivity toward DC increased to 85% (Entry 2, Table 2).
This result indicates that the presence of water in the reaction mixture affects the formation of the dicarboxymethyl derivative. Most probably the water hydrolyzes DMC forming CO2 and methanol (Scheme 2), the latter shifts the equilibrium toward the reagent or the reaction intermediates (MC1 and MC2) limiting the formation of DC.

Interestingly, repeating the synthesis of DC under anhydrous conditions and in the presence of smaller amount of K2CO3, i.e., 0.5, 0.25 mol. eq., did not affect the selectivity (Entries 3–4, Table 2) that remained in any case >85%.
A reaction has been also conducted using a catalytic amount of base, i.e. 10% mol (entry 5, Table 2). After 6 hours, the selectivity toward the DC was 90%; the monocarboxymethyl intermediates MC1 and MC2 were present only in trace.
Accurate column chromatography allowed us also to isolate one of the monocarboxymethyl derivative. Its structure was undoubtedly identified by proton nuclear magnetic resonance and resulted to be the compound labeled as MC1 that incorporates the carboxymethyl group at the exo position.
It should be mentioned that, in all the experiments conducted, the MC1 intermediate was always present in the reaction mixture in greater amount than the MC2 (see supporting information). This result can be ascribed to the different reactivity of the two hydroxyl groups present in the isosorbide molecule suggesting that the exo position is carboxymethylated more easily than the endo one.
In order to confirm this hypothesis, carboxymethylation reaction has been conducted also on isosorbide epimers, isomannide, and isoidide (Scheme 3). When isomannide was used as the starting reagent the reaction resulted slower as almost complete conversion was achieved only after 24 hours. The dicarboxymethyl isomannide resulted as the main product, although the monocarboxymethyl intermediate MCIm was also present in the relevant yield, i.e., 29% (Entry 2, Table 3).

| # | Anhydro Sugar | Time (h) | Conv. (%) | DC derivative (%) | MC derivatives |
|---|---|---|---|---|---|
| 1 | Isosorbide | 6 | 100 | 90 | MC1 8%, MC2 2% |
| 2 | Isomannide | 24 | 95 | 71 | MCIm 29% |
| 3 | Isoidide | 4 | 100 | 94 | MCIi 6% |
Note: Bold values indicate the wanted product.
Reaction conditions: isosorbide:DMC:K2CO3 1:30:0.1 equation mol.; temperature 90°C; all the reactions have been conducted in anhydrous conditions. Selectivity and conversion have been calculated by GC-MS analysis.
Conversely, isoidide resulted more reactive than isosorbide being completely converted after 4 hours at reflux temperature with almost quantitative selectivity toward the dicarboxymethyl derivative (Entry 3, Table 3).
In summary, the overall reactivity rate of the three epimers can be summarized as follows:
This result confirms that the exo group is the most reactive toward the carboxymethylation reaction as already observed in the case of isosorbide's intermediate MC1. An explanation of the observed reactivity trend could be that sterically hindered carboxymethyl group stands more comfortably at the exo position rather than at the endo one, which is obviously more constrained.Methylation of isosorbide and its epimers at reflux temperature
The methylation of isosorbide to form DMI via DMC chemistry is a complex reaction due to the presence of seven possible reaction intermediates (Scheme 4). These compounds can be divided into three categories (Scheme 4), i.e., ethers (two monomethyl derivatives MMI1 e MMI2), carboxymethyl methyl derivatives (MCE1 and MCE2), and carboxymethyl compounds (two monocarboxymethyl, MC1 e MC2 and DC).

The ether derivatives are the products of kinetically driven reactions by BAl2 mechanism, and the carboxymethyl compounds, on the other hand, are formed as a result of thermodynamic (equilibrium) reactions by a BAc2 mechanism.
The dimethyl isosorbide, as well as its reaction intermediates, can be easily identified by GC-MS analysis.
The methylation of isosorbide in batch conditions at 90°C and in the presence of a stoichiometric excess of a strong base (NaOMe) has already been reported by our group [31]. The result showed that after 24 hours, isosorbide was fully converted into DMI with a selectivity of 98%. It should be noted that in this reaction conditions, the methylation does not encompass a decarboxymethylation, but the mechanism operating in this case is BAl2 substitution on the DMC molecule [31].
In order to better understand the reactivity of isosorbide in the methylation reaction, we investigated the reactivity of its epimers isomannide and isoidide under the same reaction conditions.
Scheme 5 reports the methylation reaction of isomannide and isoidide. For both epimers there are four possible reaction intermediates (seven in the case of isosorbide) due to the more symmetrical structures.
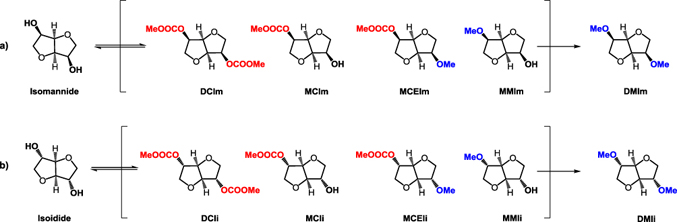
The reaction of isomannide with DMC in the presence of NaOMe resulted very effective (Scheme 5a). After five hours, GC-MS analysis showed a complete conversion of the substrate with a selectivity of 93% (Entry 2, Table 4).
| # | Anhydro Sugar | Time (h) | DM-derivate (%) | Other products (%) |
|---|---|---|---|---|
| 1 | Isosorbide | 24 | 98 | MCE1 + MCE2 2% |
| 2 | Isomannide | 5 | 93 | MMIm 7% |
| 3 | Isoidide | 24 | 61 | MMIi 4%, MCEIi 34% |
Note: Bold values indicate the wanted product.
Reaction conditions: Cyclic sugar/DMC/NaOMe 1/50/3 eq. mol.; temperature 90°C; conversion 100%. Selectivity and conversion have been calculated by GC-MS analysis.
On the other hand, isoidide resulted in the less reactive epimer (Scheme 5b). After 24 hours, despite a complete conversion of the substrate, the dimethyl isoidide was obtained only with modest yield (61%) and two reaction intermediates, namely, the monomethyl and the methyl carboxymethyl derivatives, were still present in the reaction mixture (Entry 3, Table 4).
Overall, for the methylation reaction in batch conditions, the reactivity rate of the three epimers follows the trend:
This result showed that the endo hydroxyl group is the most reactive in methylation reactions as a result of the anchimeric effect of the β-oxygen situated in the adjacent tetrahydrofuranic unit and the intramolecular hydrogen bond. Thus, the nucleophilicity of the hydroxyl in the endo position is enhanced compared to the one of the exo hydroxyl group. This explanation fits the above-reported reactivity trend.Methylation of isosorbide and its epimers in autoclave by hydrotalcite
We have previously reported that methylation of alcohols can be catalyzed by alumina or hydrotalcite operating in autoclave at high temperature. The reaction of 1-octanol and other alcohols with DMC, for instance, has been carried out in a stainless steel autoclave at 200°C resulting in the formation of the related methyl ether [22]. The methylation reaction occurs in two steps; the methylcarbonate formed in the first step undergoes decarboxylation in the second step, to give the corresponding methylether. The reactivity follows the order: primary > secondary ≫ tertiary alcohols. However, its reaction mechanism is not yet reported.
Therefore, we decided to investigate the methylation of isosorbide by adopting this reaction condition. An autoclave was charged with isosorbide, DMC (50 eq. mol.), and a stoichiometric amount (1 eq. mol.) of a base/a catalyst and was heated under pressure. Table 5 reports the data of these experiments.
| No. | Catalyst (eq. mol.) | Temp. (°C) | Press. (bar)b | Selectivity (GC-MS) % | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MMI1 | DMI | MMI2 | MC1 | MCE | MC2 | DC | ||||
| 1 | K2CO3 (1.0) | 180 | 8 | 7 | 9 | 10 | 5 | 55 | 2 | 11 |
| 2 | K2CO3 (1.0) | 200 | 12 | 4 | 57 | 7 | – | 29 | – | – |
| 3 | ButOK (1.0) | 180 | 9 | 13 | 23 | 13 | 2 | 48 | – | 1 |
| 4 | ButOK (1.0) | 200 | 12 | 5 | 55 | 6 | – | 34 | – | – |
| 5c | KW2000d | 180 | 11 | 1 | 83 | 3 | – | 12 | – | – |
| 6c | KW2000d | 200 | 20 | – | 86 | 2 | – | 12 | – | – |
| 7 | KW2000d | 200 | 20 | – | 91 | 2 | – | 7 | – | – |
Note: Bold values indicate the wanted product.
Reaction conditions: Isosorbide: DMC 1:50 eq. mol.; reaction time 24 h; conversion 100%;
The pressure increases during the proceeding of the reaction.
Reaction time 8 h.
Isosorbide:KW2000 1:1 (w/w).
When the weak base K2CO3 was used in stoichiometric amount at 180°C (Entry 1, Table 5), the substrate was completely converted after 24 hours, although the selectivity toward DMI was only scarce (9%). All of the seven reaction intermediates were present in different amount in the reaction mixture. The monocarboxymethyl methyl derivatives MCE1 and MCE2, which are not distinguishable by GC-MS analysis, were the major products.
The reaction was then repeated at higher temperature (200°C); as a result, the selectivity toward DMI increased significantly to 57%, although it was moderate.
Interestingly, using a stronger base, i.e., KOBut in the same reaction conditions at 180°C, the yield of DMI was still quite low (Entry 3, Table 5) and also at higher temperature (200°C), the selectivity of the dimethyl product was comparable to the one observed with K2CO3 (Entry 4, Table 5).
In all these latter experiments (Entries 1–4, Table 5) GC-MS analysis of the reaction mixture showed, in addition to the product of interest (DMI), the presence of different reaction intermediates. Careful column chromatography allowed us to isolate some of them. In particular, the two monomethyl derivatives MMI1 and MMI2 were recovered as pure compounds. Their structures were determined by NMR spectroscopy (see Supporting Information). The monomethyl isosorbide MMI1 has the methoxy group in exo position, whereas MMI2 is in the endo position. On the other hand, the two methyl carboxymethyl derivatives MCE1 and MCE2 were not isolable as pure, but as a mixture of the two isomers.
In order to optimize the methylation of isosorbide, it was decided to employ a different type of catalyst, i.e., hydrotalcite KW2000 (Mg0.7Al0.3O1.15), that incorporates both acidic and basic sites. The use of hydrotalcite is very attractive since, after appropriate washing and calcination, it can be recycled and reused again as reaction catalyst [65].
Hydrotalcite was calcinated at 400°C overnight prior to its use and added to a mixture of isosorbide and DMC in 1:1 w/w ratio. In these conditions, the selectivity toward DMI was almost quantitative (up to 91%) after 24 hours both at 180 °C and 200°C (Entries 5–6, Table 5, respectively). The reaction was also repeated over a period of 8 hours, giving a similar selectivity (Entry 7, Table 5).
The best-found reaction conditions have been then tested also for isosorbide epimers (Scheme 5).
Comparing the results reported in Table 5, isosorbide resulted in the most reactive substrate in autoclave conditions (Entry 1, Table 6). Isomannide (Entry 2, Table 6) and isoidide (Entry 3, Table 6) have instead shown a complete conversion only after 24 hours, with selectivity of 75% and 48%, respectively.
| No. | Sugar | Press. (bar) | Time (h) | DM-derivative (% GC-MS) | Other products (% GC-MS) |
|---|---|---|---|---|---|
| 1 | Isosorbide | 11 | 8 | 83 | MMI2 3%; MCE 12% |
| 2 | Isomannide | 10 | 24 | 75 | MCEIm 12% |
| 3 | Isoidide | 20 | 24 | 48 | MMIi 13%, MCEIi 39% |
Note: Bold values indicate the wanted product.
Reaction conditions: Sugar:DMC 1:50 eq. mol.; sugar:KW2000 1:1 (w/w). Reaction temperature 180°C; conversion 100%.
Interestingly, the reactivity rate in autoclave conditions in the presence of hydrotalcite is quite different from the one found at reflux conditions and in the presence of a base. Most probably, in the first case, the reactivity of the three epimers is influenced by the use of amphoteric catalyst hydrotalcite. In particular, although the reaction mechanism in the case of hydrotalcite has not yet been investigated in detail, we can make some general observations with respect to the catalysts that have been employed for the methylation of isosorbide.
Basic catalysts, i.e., K2CO3 and KOBut, activate the substrate, promoting the formation of the alkoxide ion, which then undergoes BAl2 attack, leading to the methyl derivative. The hydrotalcite catalytic mechanism is completely different and depends on its peculiar layered structure. In this case, most probably, DMC is activated by the metal cations present in the KW2000 structure. These cations are believed to behave as Lewis acids coordinating to the oxygen atoms of the DMC by electrostatic bonds enhancing its electrophilic character.
In order to have a better insight on the reaction mechanism, it is essential to conduct further experiments including computational calculations.
CONCLUSION
In this work, the reactivity of isosorbide and its epimers, isomannide and isoidide with DMC has been investigated in different reaction conditions in order to achieve an easy access to bio-based products by a free-halogen chemistry approach.
Carboxymethylation reaction via BAc2 mechanism has been first tested on isosorbide. The best-found conditions for DC synthesis include the use of DMC as reagent and as solvent in the presence of a catalytic amount of K2CO3 at reflux conditions. Carboxymethylation of isomannide and isoidide has also been investigated. Isomannide results in the most reactive anhydro sugar. This result shows that the exo group is more reactive; an explanation can be accounted by the bulky carboxymethyl moiety that prefers the less sterically hindered exo positions. This is also well confirmed by the higher reactivity of isomannide compared to the other epimers.
Methylation of isosorbide and its epimers has also been reported at the reflux temperature of DMC showing, in the case of base-catalyzed reaction, the higher reactivity of the endo hydroxyl group.
This result was ascribed to the anchimeric effect due to the combined effect of the β-oxygen situated in the adjacent tetrahydrofuranic unit and the intramolecular hydrogen bond present within the structure that enhances the nucleophilicity of the corresponding hydroxyl group.
However, when anphoteric catalyst hydrotalcite has been investigated in autoclave at a higher temperature, the reactivity of the epimers resulted quite different with isosorbide being the most reactive reagent.
The hydrotalcite catalytic mechanism is still not fully understood and depends on its layered structure comprising of both acidic and basic sites. Most probably acidic sites activate DMC, whereas basic sites activate the substrate. Among the catalysts investigated in autoclave conditions, hydrotalcite is very attractive since it can be easily recycled.
Our investigation on the carboxymethylation and methylation reactions in different reaction conditions allowed us also to isolate different reaction intermediates, namely, MC1, MMI1, and MMI2. The reactivity of these compounds and possibly the use of computational calculation could help in the next future to better understand the BAc2 and BAl2 mechanisms involved in the methylation of isosorbide.
It is finally noteworthy that the reported reactions allow an easy access to DMI, a bio-based high boiling green solvent and DC, a high reactive monomer for bio-based polymers. These reactions are an emblematic example of transformation of a renewable source into an industrially appealing product as they encompass free-halogen and green chemistry, and the resulting products might substitute fossil-based compounds and/or reaction intermediates.
EXPERIMENTAL
Methods
1H and 13C NMR spectra were recorded using a spectrometer Varian Unity 400 MHz in CDCl3.
The reactions carried out at high pressure were conducted in a stainless steel autoclave with a capacity of 220 mL equipped with a thermostat and a thermocouple, heated by Jacketed heating mantle, and equipped with magnetic stirring.
DMC was dried over molecular sieves (4 Å) under a nitrogen flow. K2CO3 was dried in a stove at 100°C under vacuum. Hydrotalcite KW2000 was calcined in a muffle at 400°C. All other reagents and solvents used were purchased from Sigma-Aldrich and used without further purification.
General procedure for the synthesis of dimethoxycarbonyl derivatives of isosorbide and its epimers
Example from Table 2, Entry 3. In a typical experiment, a mixture of cyclic sugar (1.00 g, 6.84 mmol), K2CO3 (0.09 g, 0.68 mmol), and DMC (18.20 g, 206.00 mmol) was placed in a 100-mL three-necked flask equipped with a refrigerant. The solution was continuously stirred heated at reflux temperature (90°C) under a constant flow of nitrogen. The course of the reaction was monitored by GC-MS, and samples were taken at regular time intervals. When the starting material was completely converted or the reaction reached equilibrium, the mixture was cooled, filtered on paper, and the solid residue washed with DMC. The solvent was evaporated under vacuum to recover the dimethoxycarbonyl derivative of the cyclic sugar.
Dimethoxycarbonyl isosorbide (DC). M.p. 51.8–54.7°C, GC-MS m/z calculated for C10H14O8 262.21, found 263.20. 1H NMR (400 MHz, CDCl3): δ = 3.80 (s, 3H), 3.82 (s, 3H), 3.86–3.95 (m, 2H), 3.99–4.09 (m, 2h), 4.54 (d, 1H), 4.89 (t, 1H), 5.05–5.10 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 55.32, 55.36, 70.69, 73.47, 76.93, 81.07, 81.45, 86.09, 155.01, and 155.33 ppm.
Dimethoxycarbonyl isomannide (DCIm). M.p. 72.5–73.9°C, GC-MS m/z calculated for C10H14O8 262.21, found 263.10. 1H NMR (400 MHz, CDCl3): δ = 3.81 (s, 6H), 3.85–3.89 (m, 2H), 4.04–4.08 (m, 2H), 4.72–4.75 (m, 2H), 4.98–5.03 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 55.38, 70.57, 76.57, 80.53, and 155.32 ppm.
Dimethoxycarbonyl isoidide (DCIi). M.p. 47.6–49.9°C calculated for C10H14O8 262.21, found 263.10. 1H NMR (400 MHz, CDCl3): δ = 3.79 (s, 6H), 3.93–4.01 (m, 4H), 4.69 (s, 2H), 5.09 (d, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 55.33, 72.53, 80.92, 85.34, and 154.91 ppm.
Monomethoxycarbonyl isosorbide (MC1) was isolated by column chromatography on silica gel, using CH2Cl2:CH3CN (8:2) as an elution mixture. GC-MS m/z calculated for C8H12O6 204.18, found 204.90. 1H NMR (400 MHz, CDCl3): δ = 2.63 (bd, 1H), 3.55–3.58 (m, 1H), 3.80 (s, 3H), 3.86–3.90 (m, 1H), 3.98–4.01 (dd, 1H), 4.12 (d, 1H), 4.30 (bq, 1H), 4.52 (d, 1H), 4.64 (t, 1H), 5.12 (d, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 55.34, 72.50, 73.48, 73.77, 81.79, 82.19, 85.59, and 154.94 ppm.
General procedure for the synthesis of dimethyl derivatives of isosorbide and its epimers
Experiment from Table 3, Entry 1. in a typical experiment, in a 100-mL three-necked flask equipped with refrigerant was prepared a mixture of cyclic sugar (1.00 g, 6.84 mmol), NaOMe (2.22 g, 41.10 mmol), and DMC (30.80 g, 342.00 mmol). The solution was heated at reflux temperature under magnetic stirring, maintaining a constant flow of nitrogen. The progress of the reaction was monitored by GC-MS on samples taken at regular time intervals until complete disappearance of the starting reagent. The mixture was then cooled, filtered on paper, and the solid residue washed with DMC. The solvent was evaporated under vacuum to recover the pure dimethyl derivatives:
Dimethyl isosorbide (DMI). GC-MS m/z was calculated for C8H14O4 174.19, found 174.10. 1H NMR (400 MHz, CDCl3): δ = 3.33 (s, 3H), 3.42 (s, 3H), 3.50–3.56 (m, 1H), 3.81–3.82 (m, 1H), 3.84–3.95 (m, 4H), 4.47 (d, 1H), 4.60 (t, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 57.31, 58.37, 69.92, 73.18, 80.09, 81.95, 86.05, 86.06 ppm.
Dimethyl isomannide (DMIm). M.p. 75.0–759°C. GC-MS m/z was calculated for C8H14O4 174.19, found 174.10. 1H NMR (400 MHz, CDCl3): δ = 3.46 (s, 6H), 3.69 (t, 2H), 3.92–3.97 (m, 2H), 4.05–4.09 (m, 2H), 4.58–4.60 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 58.52, 71.16, 80.50, and 82.20 ppm.
Dimethyl isoidide (DMIi). GC-MS m/z calculated for C8H14O4 174.19, found 174.20. 1H NMR (400 MHz, CDCl3): δ = 3.38 (s, 6H), 3.78–3.88 (m, 6H), 4.58 (s, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 57.42, 72.04, 85.12, 85.30 ppm.
By careful column cromatography in silica gel using CH2Cl2-CH3CN (9:1) as elution mixture it was also possible to isolate:
Monometyl isosorbide (MMI1). GC-MS m/z calculated for C7H12O4 160.17, found 160.10. 1H NMR (400 MHz, CDCl3): δ = 2.72 (bd, 1H), 3.37 (s, 3H), 3.53–3.57 (m, 1H), 3.82–3.87 (m, 2H), 3.90 (d, 1H), 4.03 (d, 1H), 4.26 (bq, 1H), 4.45 (d, 1H), 4.58 (t, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 57.14, 72.22, 72.93, 73.42, 81.69, 85.40, 85.60 ppm.
Monometyl isosorbide (MMI2). GC-MS m/z calculated for C7H12O4 160.17, found 160.10. 1H NMR (400 MHz, CDCl3): δ = 2.45 (s, 1H), 3.45 (s, 3H), 3.55 (t, 1H), 3.90–3.96 (m, 4H), 4.29 (m, 1H), 4.44 (d, 1H), 4.71 (t, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 58.42, 58.44, 76.02, 76.64, 79.94, 81.94, 88.55 ppm.
General procedure for the synthesis of dimethyl derivatives of isosorbide and its epimers in autoclave
Experiment from Table 4, Entry 5: In an autoclave, equipped with a magnetic stir bar, was prepared a solution with cyclic sugar (1.50 g, 10.30 mmol), hydrotalcite KW2000 (1.50 g), and DMC (46.40 g, 515.00 mmol). The autoclave was hermetically sealed under a nitrogen atmosphere and then heated to 180°C, reaching a pressure of ca 15 bar. The progress of the reaction was monitored by GC-MS. Once the starting sugar was fully converted, the mixture was then cooled, filtered on paper, and the solid residue washed with DMC. The solvent was evaporated under vacuum to obtain the methyl derivative of the cyclic sugar according to the previously reported procedure.