- Record: found
- Abstract: found
- Article: not found
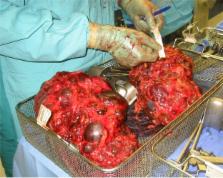
Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment
review-article
Read this article at
ScienceOpenPMC
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Both autosomal dominant and recessive polycystic kidney disease are conditions with severe associated morbidity and mortality. Recent advances in the understanding of the genetic and molecular pathogenesis of both ADPKD and ARPKD have resulted in new, targeted therapies designed to disrupt cell signaling pathways responsible for the abnormal cell proliferation, dedifferentiation, apoptosis, and fluid secretion characteristic of the disease. Herein we review the current understanding of the pathophysiology of these conditions, as well as the current treatments derived from our understanding of the mechanisms of these diseases.
Related collections
Most cited references140
- Record: found
- Abstract: found
- Article: not found
Autosomal dominant polycystic kidney disease.
Vicente Torres, Peter Harris, Yves Pirson (2007)
- Record: found
- Abstract: found
- Article: not found
Unified criteria for ultrasonographic diagnosis of ADPKD.
York Pei, James Obaji, Annie Dupuis … (2009)
- Record: found
- Abstract: found
- Article: not found
Volume progression in polycystic kidney disease.
Jared J Grantham, Vicente Torres, Arlene Chapman … (2006)