- Record: found
- Abstract: found
- Article: found
Statistical Quantification of Methylation Levels by Next-Generation Sequencing
Read this article at
Abstract
Background/Aims
Recently, next-generation sequencing-based technologies have enabled DNA methylation profiling at high resolution and low cost. Methyl-Seq and Reduced Representation Bisulfite Sequencing (RRBS) are two such technologies that interrogate methylation levels at CpG sites throughout the entire human genome. With rapid reduction of sequencing costs, these technologies will enable epigenotyping of large cohorts for phenotypic association studies. Existing quantification methods for sequencing-based methylation profiling are simplistic and do not deal with the noise due to the random sampling nature of sequencing and various experimental artifacts. Therefore, there is a need to investigate the statistical issues related to the quantification of methylation levels for these emerging technologies, with the goal of developing an accurate quantification method.
Methods
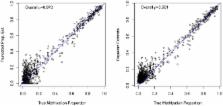
In this paper, we propose two methods for Methyl-Seq quantification. The first method, the Maximum Likelihood estimate, is both conceptually intuitive and computationally simple. However, this estimate is biased at extreme methylation levels and does not provide variance estimation. The second method, based on Bayesian hierarchical model, allows variance estimation of methylation levels, and provides a flexible framework to adjust technical bias in the sequencing process.
Results
We compare the previously proposed binary method, the Maximum Likelihood (ML) method, and the Bayesian method. In both simulation and real data analysis of Methyl-Seq data, the Bayesian method offers the most accurate quantification. The ML method is slightly less accurate than the Bayesian method. But both our proposed methods outperform the original binary method in Methyl-Seq. In addition, we applied these quantification methods to simulation data and show that, with sequencing depth above 40–300 (which varies with different tissue samples) per cleavage site, Methyl-Seq offers a comparable quantification consistency as microarrays.
Related collections
Most cited references21
- Record: found
- Abstract: found
- Article: not found
Principles and challenges of genomewide DNA methylation analysis.
- Record: found
- Abstract: found
- Article: not found
Genomic mapping by fingerprinting random clones: a mathematical analysis.
- Record: found
- Abstract: found
- Article: not found