- Record: found
- Abstract: found
- Article: found
tRNA Methyltransferase Homolog Gene TRMT10A Mutation in Young Onset Diabetes and Primary Microcephaly in Humans
Read this article at
Abstract
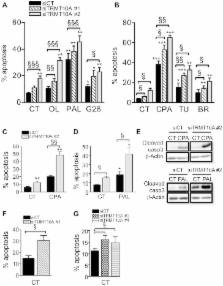
We describe a new syndrome of young onset diabetes, short stature and microcephaly with intellectual disability in a large consanguineous family with three affected children. Linkage analysis and whole exome sequencing were used to identify the causal nonsense mutation, which changed an arginine codon into a stop at position 127 of the tRNA methyltransferase homolog gene TRMT10A (also called RG9MTD2). TRMT10A mRNA and protein were absent in lymphoblasts from the affected siblings. TRMT10A is ubiquitously expressed but enriched in brain and pancreatic islets, consistent with the tissues affected in this syndrome. In situ hybridization studies showed that TRMT10A is expressed in human embryonic and fetal brain. TRMT10A is the mammalian ortholog of S. cerevisiae TRM10, previously shown to catalyze the methylation of guanine 9 (m 1G 9) in several tRNAs. Consistent with this putative function, in silico topology prediction indicated that TRMT10A has predominant nuclear localization, which we experimentally confirmed by immunofluorescence and confocal microscopy. TRMT10A localizes to the nucleolus of β- and non-β-cells, where tRNA modifications occur. TRMT10A silencing induces rat and human β-cell apoptosis. Taken together, we propose that TRMT10A deficiency negatively affects β-cell mass and the pool of neurons in the developing brain. This is the first study describing the impact of TRMT10A deficiency in mammals, highlighting a role in the pathogenesis of microcephaly and early onset diabetes. In light of the recent report that the type 2 diabetes candidate gene CDKAL1 is a tRNA methylthiotransferase, the findings in this family suggest broader relevance of tRNA methyltransferases in the pathogenesis of type 2 diabetes.
Author Summary
The inherited predisposition to type 2 diabetes is attributed to common variants in over 60 loci. Among these risk variants is CDKAL1, which has recently been shown to be a tRNA modifying enzyme (methylthiotransferase). Genetic variants of different severity can generate a spectrum of monogenic and polygenic forms of diabetes. Here we describe a new syndrome of young onset diabetes, short stature and microcephaly (small brain size) with intellectual disability in a large consanguineous family. By linkage analysis and whole exome sequencing we identified a nonsense mutation in TRMT10A, a gene that has hitherto not been studied in mammals. The yeast homolog TRM10 has been shown to be a tRNA modifying enzyme with methyltransferase activity. We demonstrate that TRMT10A mRNA and protein are absent in cells from the affected siblings. TRMT10A localizes to the nucleolus, where tRNA modifications occur. TRMT10A silencing induces cell death in insulin-producing pancreatic β-cells, suggesting that TRMT10A deficiency may reduce β-cell mass and the pool of neurons in the brain. This is the first study describing the impact of TRMT10A deficiency in man. Our findings may have broader relevance for the understanding of the pathogenesis of type 2 diabetes and microcephaly.
Related collections
Most cited references47
- Record: found
- Abstract: found
- Article: not found
The nonsense-mediated decay RNA surveillance pathway.
- Record: found
- Abstract: found
- Article: not found
Six classes of nuclear localization signals specific to different binding grooves of importin alpha.
- Record: found
- Abstract: found
- Article: not found