- Record: found
- Abstract: found
- Article: found
Mutations that are a common cause of Leber congenital amaurosis in northern America are rare in Southern India
Read this article at
Abstract
Purpose
To test patients from southern India for the presence of mutations that most commonly cause Leber congenital amaurosis (LCA) in northern America.
Methods
A review of the literature identified 177 unique LCA causing mutations in eight different genes: aryl hydrocarbon receptor interacting protein-like 1 ( AIPL1), crumbs homolog 1 ( CRB1), cone-rod homeobox ( CRX), guanylate cyclase 2D ( GUCY2D), nephronophthisis 6 ( NPHP6), retinol dehydrogenase 12 ( RDH12), retinal pigment epithelium-specific protein 65 kDa ( RPE65), and retinitis pigmentosa GTPase regulator interacting protein 1 ( RPGRIP1). Allele-specific ligation assay and bidirectional sequencing were used to test 38 unrelated LCA patients from southern India for 104 of these mutations, which contribute to more than 30% of the LCA cases in a northern American population.
Results
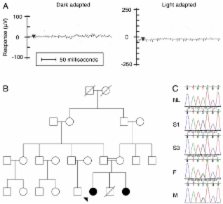
Only one participant was found to harbor one of the 104 mutations in the allele-specific assay (homozygous RPE65 Tyr368His). A mutation that was not part of the assay (homozygous RPE65 Tyr143Asp) was incidentally detected in a second patient when an equivocal signal from one allele on the assay was followed up with automated DNA sequencing.
Conclusions
Mutations that contribute to 30% of the LCA cases in northern America were detected in only 2.6% of LCA cases in our cohort from southern India. There were no instances of IVS26 c.2991+1655 A>G in NPHP6, the most commonly detected mutation in LCA. These data suggest that LCA in India is caused primarily by a different set of mutations in the same genes associated with disease in northern America, or by mutations in other genes that have not yet been discovered. Therefore, mutation-specific assays developed for European and northern American cohorts may not be suited for testing LCA patients from India or other ethnically distinct populations.
Related collections
Most cited references15
- Record: found
- Abstract: found
- Article: not found
Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy.
- Record: found
- Abstract: found
- Article: not found
Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12).
- Record: found
- Abstract: found
- Article: not found