- Record: found
- Abstract: found
- Article: found
Successful Recovery of Nuclear Protein-Coding Genes from Small Insects in Museums Using Illumina Sequencing
Read this article at
Abstract
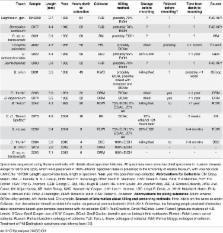
In this paper we explore high-throughput Illumina sequencing of nuclear protein-coding, ribosomal, and mitochondrial genes in small, dried insects stored in natural history collections. We sequenced one tenebrionid beetle and 12 carabid beetles ranging in size from 3.7 to 9.7 mm in length that have been stored in various museums for 4 to 84 years. Although we chose a number of old, small specimens for which we expected low sequence recovery, we successfully recovered at least some low-copy nuclear protein-coding genes from all specimens. For example, in one 56-year-old beetle, 4.4 mm in length, our de novo assembly recovered about 63% of approximately 41,900 nucleotides in a target suite of 67 nuclear protein-coding gene fragments, and 70% using a reference-based assembly. Even in the least successfully sequenced carabid specimen, reference-based assembly yielded fragments that were at least 50% of the target length for 34 of 67 nuclear protein-coding gene fragments. Exploration of alternative references for reference-based assembly revealed few signs of bias created by the reference. For all specimens we recovered almost complete copies of ribosomal and mitochondrial genes. We verified the general accuracy of the sequences through comparisons with sequences obtained from PCR and Sanger sequencing, including of conspecific, fresh specimens, and through phylogenetic analysis that tested the placement of sequences in predicted regions. A few possible inaccuracies in the sequences were detected, but these rarely affected the phylogenetic placement of the samples. Although our sample sizes are low, an exploratory regression study suggests that the dominant factor in predicting success at recovering nuclear protein-coding genes is a high number of Illumina reads, with success at PCR of COI and killing by immersion in ethanol being secondary factors; in analyses of only high-read samples, the primary significant explanatory variable was body length, with small beetles being more successfully sequenced.
Related collections
Most cited references42

- Record: found
- Abstract: found
- Article: found
Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform
- Record: found
- Abstract: found
- Article: not found
The genome of the model beetle and pest Tribolium castaneum.
- Record: found
- Abstract: found
- Article: not found