- Record: found
- Abstract: found
- Article: found
Enpp1 is an anti-aging factor that regulates Klotho under phosphate overload conditions
Read this article at
Abstract
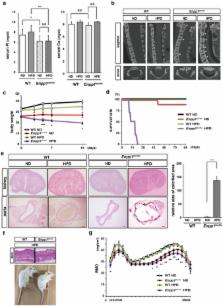
Control of phosphate metabolism is crucial to regulate aging in mammals. Klotho is a well-known anti-aging factor that regulates phosphate metabolism: mice mutant or deficient in Klotho exhibit phenotypes resembling human aging. Here we show that ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1) is required for Klotho expression under phosphate overload conditions. Loss-of-function Enpp1 ttw/ttw mice under phosphate overload conditions exhibited phenotypes resembling human aging and Klotho mutants, such as short life span, arteriosclerosis and osteoporosis, with elevated serum 1,25(OH) 2D 3 levels. Enpp1 ttw/ttw mice also exhibited significantly reduced renal Klotho expression under phosphate overload conditions, and aging phenotypes in these mice were rescued by Klotho overexpression, a low vitamin D diet or vitamin D receptor knockout. These findings indicate that Enpp1 plays a crucial role in regulating aging via Klotho expression under phosphate overload conditions.
Related collections
Most cited references54
- Record: found
- Abstract: found
- Article: not found
Klotho deficiency causes vascular calcification in chronic kidney disease.
- Record: found
- Abstract: found
- Article: not found
Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23.
- Record: found
- Abstract: found
- Article: not found