- Record: found
- Abstract: found
- Article: not found
Mitochondrial Regulation of Store-operated Calcium Signaling in T Lymphocytes
Read this article at

Abstract
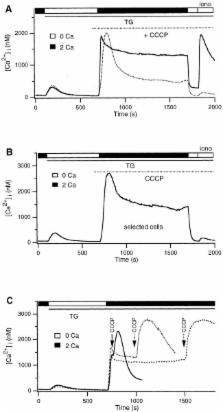
Mitochondria act as potent buffers of intracellular Ca 2+ in many cells, but a more active role in modulating the generation of Ca 2+ signals is not well established. We have investigated the ability of mitochondria to modulate store-operated or “capacitative” Ca 2+ entry in Jurkat leukemic T cells and human T lymphocytes using fluorescence imaging techniques. Depletion of the ER Ca 2+ store with thapsigargin (TG) activates Ca 2+ release-activated Ca 2+ (CRAC) channels in T cells, and the ensuing influx of Ca 2+ loads a TG- insensitive intracellular store that by several criteria appears to be mitochondria. Loading of this store is prevented by carbonyl cyanide m-chlorophenylhydrazone or by antimycin A1 + oligomycin, agents that are known to inhibit mitochondrial Ca 2+ import by dissipating the mitochondrial membrane potential. Conversely, intracellular Na + depletion, which inhibits Na +-dependent Ca 2+ export from mitochondria, enhances store loading. In addition, we find that rhod-2 labels mitochondria in T cells, and it reports changes in Ca 2+ levels that are consistent with its localization in the TG-insensitive store. Ca 2+ uptake by the mitochondrial store is sensitive (threshold is <400 nM cytosolic Ca 2+), rapid (detectable within 8 s), and does not readily saturate. The rate of mitochondrial Ca 2+ uptake is sensitive to extracellular [Ca 2+], indicating that mitochondria sense Ca 2+ gradients near CRAC channels. Remarkably, mitochondrial uncouplers or Na + depletion prevent the ability of T cells to maintain a high rate of capacitative Ca 2+ entry over prolonged periods of >10 min. Under these conditions, the rate of Ca 2+ influx in single cells undergoes abrupt transitions from a high influx to a low influx state. These results demonstrate that mitochondria not only buffer the Ca 2+ that enters T cells via store-operated Ca 2+ channels, but also play an active role in modulating the rate of capacitative Ca 2+ entry.
Related collections
Most cited references59
- Record: found
- Abstract: found
- Article: not found
Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function.
- Record: found
- Abstract: found
- Article: not found