- Record: found
- Abstract: found
- Article: not found
Tubular Epithelial and Peritubular Capillary Endothelial Injury in COVID-19 AKI
case-report

John C. Papadimitriou , M.D, Ph.D.
∗∗ ,
Cinthia B. Drachenberg , M.D.,
David Kleiner , M.D.
∗ ,
Nadia Choudhri , M.D.
# ,
Abdolreza Haririan , M.D.
# ,
Valeriu Cebotaru , M.D.
#
5 November 2020
Read this article at

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
INTRODUCTION
Multiple reports describe the respiratory system involvement in coronavirus disease
2019 (COVID-19), but in patients that require hospitalization concurrent renal dysfunction
is common
1
. In a large study, more than 36% of patients developed acute kidney injury (AKI),
and of those, 14.3% required renal replacement therapy (RRT) S1. Development of AKI
occurs early, is temporally associated with acute respiratory failure, and carries
worse overall prognosis S1-4.
The etiology of AKI in COVID-19 is considered to be multifactorial, due to volume
depletion, poor renal perfusion, sepsis, and systemic inflammatory cytokine storm
S5. SARS-CoV-2 renal tropism has been suggested and significant amounts of viral RNA
were detected by PCR in kidney tissue from some patients with viremia S6 . Conclusive
morphological evidence of SARS-CoV-2 viral particles in the renal parenchyma is lacking,
although electron microscopy (EM) studies have shown abundant intracellular vesicular
structures, resembling SARS-CoV-2 viral particles
2
. These structures, most likely representing clathrin-coated transport vesicles
3
, do not fulfill the morphological criteria for corona virions S8-11 , but highlight
a profound ultrastructural alteration in renal tissue, comparable to that seen in
other cell types after oxidative stress injury
4
,
5
.
In the lung, vascular involvement, including endothelial cell damage, vascular inflammation,
thrombosis and microangiopathy, as well as regeneration with neoangiogenesis, points
to the vascular endothelium as an important target of COVID-19 S7.8,12,13. In the
kidney, severe vascular congestion and possible microthrombi were interpreted as evidence
of vascular injury
6
, however, systematic ultrastructural evaluation of the renal endothelium in COVID-19
is not available.
CASE 1
A 52 year old man with longstanding well controlled HIV, HTN, CAD, and Factor V deficiency,
presented to the emergency department with severe vomiting and diarrhea for about
a week, and tested positive for a nasal swab RT-PCR for SARS-CoV-2. He also noted
episodes of epistaxis and myalgias, but denied fever, chills, cough, shortness of
breath, chest pain or edema. His blood pressure was 120-140s/80s, and he was not hypoxic.
Initial BUN and serum creatinine were 30mg/dL and 7.5mg/dL, respectively. Serum creatinine
was normal 2 months earlier. CBC was normal, except for mild normocytic anemia. HIV
viral load was undetectable and CD4 count was normal. Ferritin level was high (1427
ng/mL, normal 23-336), as were CRP (109 mg/L, normal <6) and D-dimer (630 ng/mL, normal
0-500). The patient was initiated, on intravenous fluids and admitted to the intensive
care unit. He was initially alert and conversant, but notably anxious with no other
significant clinical findings. He developed severe epistaxis and subsequently acute
hypoxemic respiratory failure requiring intubation and mechanical ventilation on day
2. Imaging studies of the lungs showed pulmonary edema. Piperacillin/tazobactam and
hydroxychloroquine were initiated, as well as renal replacement therapy (RRT), due
to persistent azotemia. The clinical course was complicated by paroxysmal atrial fibrillation
treated with amiodarone, and acute anemia and thrombocytopenia requiring transfusion
of red blood cells and platelets. After 48 hours of mechanical ventilation the patient
was extubated and was stable on room air. Gastrointestinal symptoms, including nausea
and diarrhea, persisted for few days. Spot urine protein/creatinine ratio revealed
1.85g/g proteinuria. C3 and C4 complement components were normal, and serological
studies including autoimmune and hepatitis panels were negative. There was no evidence
of monoclonal gammopathy. Renal biopsy was performed on day 10 of hospital stay for
proteinuria and lack of improvement of the renal function. Nasal swabs for SARS-CoV-2,
repeated 7 and 14 days after admission were negative. During and after hospitalization
the patient had improving urine output, but required intermittent hemodialysis. RRT
was stopped 11 weeks after the COVID-19 diagnosis.
Case 2
A 64 year-old man with history of atrial fibrillation, on home aspirin, hyperlipidemia
and gout, presented with cough, fever, and chest pain. A nasal swab RT-PCR for SARS-CoV-2
was positive. He was admitted for hypoxemia and on day five was intubated due to worsening
hypoxemia. He was started on intravenous heparin anticoagulation for atrial fibrillation
and then transitioned to apixaban. His hospital stay was complicated by a large volume
hematemesis and coffee ground emesis requiring 4 units of blood and plasma transfusions
and was started on Norepinephrine for hypotension. He then developed atrial fibrillation
with rapid ventricular response and was started on Amiodarone infusion and Diltiazem
drip. Two attempts at cardioversion were not successful. His sputum grew E. coli and
he was started on Meropenem on hospital day 13. On hospital day 19 the patient was
found to have a decreased mental status and had a computerized tomography of the head,
that did not show any acute process.
On day 22, the patient underwent a tracheostomy and on the same day he was started
on continuous RRT for persistent azotemia and volume removal. On hospital day 29 he
was transitioned to intermittent hemodialysis. On hospital day 33 hemodialysis was
stopped. He came off ventilator support on day 78. His hospital course was complicated
by MRSA bacteremia treated with Linezolid, pseudomonas and E. Coli ventilator associated
pneumonia treated with intravenous Imipenem/cilastatin/relebactam and inhaled colistin
and polymyxin B. He also developed a right axillary and right subclavian deep venous
thrombosis and was started on intravenous heparin.
Initial laboratory tests revealed BUN 20 mg/dL, creatinine 1.4 mg/dL, WBC 4200/mcL,
Hb 13.4 g/dL, platelets 131K/mcL, INR 1.0, AST 93 unit/L, ALT 83 unit/L. His baseline
creatinine was 1.0 mg/dL. BUN and creatinine peaked at 130 mg/dl and 4.31 mg/dL, respectively.
D-Dimer peaked at 15,230 ng/mL, CRP peaked at 31.3 ng/mL and persistently high Ferritin
reached 4,461.7 ng/mL at day 22 (normal 23-336). Nasal swabs for SARS-CoV-2, 10 and
17 days after admission were negative
On hospital day 81 the patient was found to have 7.4 gm/gm of protein on urine spot
protein to creatinine ratio. Twenty four-hour urine collection revealed 4.37 gm of
proteinuria. Autoimmune and hepatitis serological studies were negative and there
was no evidence of monoclonal gammopathy. Renal biopsy was performed on hospital day
84. On biopsy day his BUN and creatinine were 51 mg/dL and 1.58 mg/dL, respectively,
with an eGFR of 36mL/min (eGFR at admission was >60).
RENAL BIOPSY FINDINGS
Light microscopy
Marked and diffuse tubular cell injury was seen in both biopsies, with involvement
of all cortical tubular segments. The day 10 biopsy (D10Bx) from patient 1 showed
severe diffuse simplification of the tubular epithelium, with marked cytoplasmic blebbing-vacuolization,
loss of cell polarity, loss of brush border and spotty or confluent cell drop-out
(Fig. 1
A). There were also prominent protein casts and areas of tubular cell sloughing (Suppl
Fig. 1A-D). The day 84 biopsy (D84Bx) from patient 2 showed diffuse tubular injury
characterized by cytoplasmic swelling with marked coarse and isometric vacuolization
(Fig. 1B). Also noted was irregular simplification with partial loss of brush border
of the proximal tubules, admixed with hyperplastic and reparative changes in all tubule
segments (Fig. 1B). Dense protein and cellular casts were noted in both cases. (Suppl
Figures 1D and 2A)
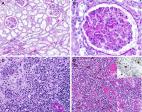
FIGURE 1
Tubular injury. A: D10Bx Tubular epithelium with marked cytoplasmic vacuolization,
blebbing, loss of brush border and spotty cell drop-out. The PTC show endothelial
cell changes, including nuclear enlargement and protrusion into the lumen. There is
also perivascular and luminal accumulation of mononuclear cells (arrow). Also see
Supplemental Figure 2D. B: D84Bx Tubular epithelium with diffuse cell injury, cytoplasmic
swelling, vacuolization and blebbing. There is loss of cell polarity, irregular simplification
and loss of brush border admixed with marked reparative changes (center and right).
Abnormal PTC endothelial cell lining (arrows) with nuclear enlargement and hyperchromasia.
Also see Supplemental Figure 2D. C: D10Bx Electron micrograph of tubule with severe
tubular epithelial cell injury with cell sloughing and denudation of the basement
membrane. The nuclei appear pyknotic and the cytoplasm severely vacuolated. Fragments
of membranes appear in the lumen (arrow) D: D84Bx, Cytoplasmic dissolution and widespread
densities consistent with damaged phospholipids suggestive of oxidative membrane injury
(arrow heads). The mitochondria appear mostly condensed. E: D10Bx, Disintegration
of the brush border and extensive cytoplasmic vesiculation. The mitochondria appear
markedly swollen or condensed, with clusters of small mitochondria (mitospheres) (arrow).
Bars: A,B 25 microns, C 3 microns, D,E 2 microns
The peritubular capillaries (PTC) were prominent in both biopsies. There was peritubular
capillary dilatation with endothelial cell nuclear enlargement and luminal protrusion
(FIGURE 1, FIGURE 2A,D). Interstitial inflammation was sparse on routine stains, but
the CD68 immunostain highlighted clusters of monocytes/macrophages, predominantly
around and in peritubular capillaries in both biopsies (Fig. 2 C and 2F). CD31 stains
for evaluation of the microvasculature showed disturbed and severely diminished staining
consistent with rupture or disintegration/lysis of most PTC endothelial cells in the
D10Bx (Fig. 2B), whereas the stain strongly highlighted the enlarged and prominent
endothelial PTC endothelial cell lining in the D84Bx (Fig. 2E).
FIGURE 2
Endothelial injury D10Bx A: PTC (arrow) are prominent due to luminal dilatation, and
nuclear enlargement and hyperchromasia of the endothelial cells. There are intraluminal
and perivascular mononuclear cell infiltrates. B: CD31 immunostain is weak and highlights
cytoplasmic vesiculation and dissolution of the PTC endothelium (arrows). C: CD68
immunostain highlights accumulation of macrophages in PTC areas. D84Bx D: PTC (arrow)
are irregularly prominent due to enlargement of the endothelial cell nuclei and increase
in mononuclear cells within lumina and surrounding interstitium. The lumina are narrowed
in several instances. E: CD31 highlights the PTC with markedly swollen, hyperplastic
endothelial cells. F: CD68 immunostain highlights macrophages in and around PTC. Also
see Supplemental Figure 5 for AKI without features of OSI. Bar: A-F 20 microns
On light microscopy the glomeruli were essentially normal with only minimal increase
in mesangial matrix on the D10BX (Suppl Fig. 1A) and showed perihilar focal segmental
glomerulosclerosis (secondary) on the D84Bx (Fig. 2B). Chronic changes were insignificant
in both biopsies, with the trichrome stain showing only mild interstitial fibrosis
and tubular atrophy (≈10-15% of the cortical areas). The arteries were widely patent,
and thrombotic/microangiopathic features were absent. Routine immunofluorescence studies
including IgG, IgM, IgA, C3, C4 and C1q, as well as immunohistochemical stains for
C3d and C4d stain were negative in both biopsies.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay (TUNEL) for evaluation
of apoptosis showed only rare apoptotic cells (0-4 apoptotic cells per 400x field)
in both biopsies.
ISH for SARS-CoV-2 spike and nuclear capside RNA (RNAscope) were negative in both
biopsies.
ULTRASTRUCTURAL FINDINGS
Tubular epithelial cell injury was extensive in both biopsies, with more prominent
sloughing and denudation of the tubular basement membranes seen on the D10Bx (Fig.
1C). In both samples the tubular epithelial cells displayed large areas of cytoplasmic
vacuolization with abnormal/disintegrating brush border due to fragmentation or vesiculation
of its membranes (Fig. 1 C,E, Suppl Fig. 3B). Overall, there were extensive vesicular
changes with abundant clathrin-coated vesicles admixed with smooth walled vesicles
(Fig. 3
C,E,F). The mitochondria were markedly abnormal, with a wide range of changes including
matrix condensation, or swelling, dissolution of cristae, myelin figure formation
and accumulation of flocculent densities (Fig. 3 A,B,C, and D, Suppl. Fig 3A). Additionally
the mitochondria displayed marked size variation with numerous small spherical mitochrondria
(mitospheres) (Fig. 3B) S14. Disarray of the cytoskeleton with accumulation of collapsing
bundles of intermediate and thin filaments was noted in the most swollen tubular cells
(Fig. 3D) S15.
FIGURE 3
Tubular Epithelial Cell Changes. A: D10Bx Tubular epithelial cell shows marked cytoplasmic
membrane vesiculation: large vacuoles with densities also seen (arrows). Condensed
mitochondria (arrowheads). B: D84Bx Marked mitochondrial swelling and/or condensation,
occasionally both within the same mitochondrion (segmentation, arrows). Occasional
small mitochondria (mitospheres) also noted. Flocculent densites present in rare mitochondria
(arrowhead).C D10Bx Marked vesiculation of the cytoplasm including small smooth walled
vesicles (asterisk) and a multivesicular body to the left. A large myelin figure (arrows)
located in the vicinity of atypically shaped condensed mitochondria. D D84Bx Marked
myelin figure formation including myelin containing mitochondria (asterisk). Cytoskeletal
filament collapse noted (arrows). E D10Bx Tubular cells show marked vesiculation of
the cytoplasm, including large vesicles and abundant clathrin-coated vesicles resembling
viral particles (arrows). F D10Bx, tubular epithelial cell cytoplasm with large numbers
of vesicles coated by clathrin resembling coronavirus spikes. Insert: Clathrin-coated
invagination of the plasma membrane. Bars: A,D 1 micron, B 2 microns, C 400nm, E 600nm,
F 300nm (Insert 100nm)
On EM, the PTC on the D10Bx showed very severe endothelial injury and/or rupture (Fig
4
A,B). The endothelial cell cytoplasmic membranes and the organelles showed marked
vesiculation and dissolution. The mitochondria appeared condensed and engulfed by
segments of rough endoplasmic reticulum, a feature consistent with autophagy (Fig.
4A). On the D84Bx, the PTC were consistently abnormal with swollen or activated appearing
endothelial cells and infiltrating monocytes and lymphocytes (Fig. 4C and D). The
mitochondrial changes were similar to the D10Bx. In addition, there was diffuse, extensive
multilamellation of their basal laminae, up to 8 layers (Fig. 4 E and F), consistent
with endothelial cell injury and regeneration.
FIGURE 4
Endothelial Cell Changes. D10Bx A: PTC endothelial cell with marked cytoplasmic vesiculation/dissolution.
Condensed mitochondria wrapped by segments of RER (arrow). B: PTC endothelial cell
with marked membrane injury and massive vesiculation. Abundant cellular fragments
shedding into the lumen. D84Bx C: PTC with swelling and hypertrophy of endothelial
cells. D: PTC with swollen endothelial cell (E). The lumen is distended by monocytes
(arrows) and a lymphocyte (arrowhead). E: PTC with fragmented endothelial cell lining
(arrow). The basal lamina is multilamellated. F: Segment of peritubular capillary
wall with marked basal lamina multilamellation (arrow). An abnormal endothelial cell
protrudes towards the lumen showing rough endoplasmic reticulum wrapping mitochondria
suggestive of autophagy.Bars: A,E and F 1 micron, B,C and D 2 microns. Also see Suppl
Fig 5 for AKI without OSI.
Glomerular endothelial cells and podocytes showed focal cell swelling and focal accumulation
of clathrin-coated or smooth vesicles, however, the foot processes of podocytes were
largely preserved in both biopsies (Supl Fig 4).
Extensive accumulation of clathrin-coated vesicles with protruding spikes towards
the cytoplasm resembling coronavirus in some instances, were observed in both biopsies
but were more prominent in the D10Bx (Fig. 3E and F). True, viral particles were not
identified in either biopsy.
DISCUSSION
COVID-19 may be minimally symptomatic, or present with severe involvement of multiple
organ systems. Pneumonia, the most common manifestation of SARS-CoV-2 infection, occurs
after engagement of the virus with the ACE2 receptor expressed in type II pneumocytes.
An abundance of ACE2 receptors in other cell types, including renal tubular cells,
enterocytes and endothelial cells, can explain some of the other manifestations of
COVID-19 S16. Several studies have pointed to the vascular endothelium as an important
target of COVID-19 pathophysiology in several organs, e.g. heart and lung S8,12 .
Endothelial injury in these circumstances can lead to recruitment of immune cells,
complement activation and potentially thrombosis S17.
Renal involvement in the form of acute kidney injury (AKI) carries worse prognosis
in COVID-19, and is an increasingly recognized complication in patients with severe
disease and in patients with pre-existing conditions S2,7,18-2114. In addition to
AKI, 40% of the patients have 2-3+ proteinuria, leukocyturia and/or hematuria S1.
Severe multiorgan involvement in COVID-19 is generally attributed to a dysregulated
host response, initially triggered by innate immune mechanisms upon encounter with
the virus. An aggressive and exaggerated hyper-inflammatory reaction leads to ongoing
release of pro-inflammatory mediators, including abundant cytokines, that cause further
host tissue damage. The amplified chain reaction results in the syndrome of viral
“sepsis”
7
. Morphological studies of AKI in sepsis overall, as well as in hyper-inflammatory
reactions, needed to validate the above hypothesis are, however, very limited S22.
The morphological findings in the two patients presented here are highly consistent
with damage induced by oxidative stress/injury (OSI) secondary to hyperinflammation,
in both tubular and endothelial cells. This type of cell injury is characterized by
severe, diffuse damage to cellular membranes, leading to microvesiculation and dissolution
of the latter, as well as prominent formation of myelin figures. Damage to mitochondrial
structure and function is also very characteristic, as is the accumulation of abundant
cytoplasmic transport vesicles both clathrin- coated as well as smooth walled 4,5
S11,23 . Although complement mediated injury has been proposed to play a role in COVID-19
S17, we did not observe in these cases morphologically or immunohistochemically typical
features associated with complement mediated cell injury, which is generally characterized
by nuclear/cytoplasmic and mitochondrial changes consistent with ion and fluid deregulation,
rather than by features of OSI S24. Similarly, significant degree of apoptosis, that
is a common form of cell loss in ischemic injury S25, was not significant either morphologically
of by TUNEL studies.
Tubular cell ultrastructural morphology in these two cases was also different from
those characterizing the most common forms of acute tubular injury extensively described
by Olsen et al. S26,27
Based on these limited studies, we suggest that OSI can play a very significant role
in AKI in COVID-19. OSI is a common pathway of cell injury, resulting from a variety
of processes, several of which can be identified in systemic viral infection in general,
and in COVID-19 specifically.
There appear to be several similarities between overall sepsis induced AKI and COVID-19
AKI, including early hemodynamic changes, leading to oxidant agent generation, especially
in the peritubular capillary miroenvironment
8
. Respiratory viruses in general, are associated with cytokine production/storm and
OSI, leading to cell injury and death
9
, an association that could be relevant in the pathophysiological scenario of sepsis/COVID-19
related AKI.
A dysregulated inflammatory response includes overactivated macrophages and potentially
neutrophils, producing a cytokine storm that is followed by a “free radical storm”
S22,28. Both of our cases, as well as other studies, have shown the prominence of
macrophages/monocytes in COVID-19 S29,30 . Furthermore, alterations in the iron metabolism,
including markedly increased serum ferritin that are characteristic of severe COVID-19,
also seen in our two patients, can contribute to the generation of free radicals and
OSI S31.
More specifically, hyperferritenimia can lead to widespread tissue injury through
massive and uncontrolled activation of T-lymphocytes and of macrophages, followed
by excessive production of inflammatory cytokines S32,33. This mechanism is similar
to the one of some challenging rheumatic diseases, characterized by hyperferritenemia,
high mortality, macrophage activation and multiple organ dysfunction S32. Macrophages
can release iron through the action of ferroportin, a process that can be blocked
by hepcidin S34. Interestingly, hepcidin or hepcidin-like activity is markedly increased
in COVID-19, potentially leading to entrapment of iron within cells-particularly macrophages,
thus further contributing to the vicious cycle of cytokine-free radical storms S31,34-36.
Severe or protracted COVID-19 AKI might be pathophysiologically similar to other entities
covered under the general “hyperferritenemic syndrome” umbrella, that also includes
septic shock S37,38. The morphological results in the two presented cases are different
from the typical features of classical ATN (ischemic or toxic), that typically presents
with less impressive tubular epithelial cell ultrastructural damage, mainly characterized
by diminution of the brush border and basolateral infoldings S26,27. In contrast,
the presence of generalized membrane injury, marked mitochondrial changes and finally
the prominence of clathrin-coated “viral-like” transport vesicles, are most consistent
with OSI. Furthermore, the observed prominent and similar damage in both epithelial
and endothelial cell types is a finding that supports a generalized injurious process
such as OSI 4,5 S11,39-45 . This pathogenetic mechanism has also been implicated in
the pathogenesis of sepsis induced AKI in general, but scarcity of biopsy material
in this clinical context has hindered morphological and clinical correlations S38.
In the COVID-19 AKI context, an additional feature of particular interest, suggesting
protracted endothelial injury and repair in the PTC, is the observed multilamellation
of the endothelial basal cell lamina, observed in the late biopsy (D84Bx). This change
is indicative of repeated endothelial cell injury, regeneration and repair, and is
reminiscent of the PTC response induced from ongoing injury and remodeling seen in
antibody mediated allograft rejection. The OSI pattern of cellular injury is, however,
not identified in tubules and endothelium in antibody mediated rejection S46, suggesting
a different form of initial insult by a similar repair pathway.
The proposed contribution of oxidative stress damage in COVID-19 patients, could account
for the increased morbidity and mortality in patients with pre-existing conditions
e.g. older age, diabetes, obesity or hypertension. All of these conditions are characterized
by cumulative oxidative damage and weaker defenses against it
9
.
Related collections
Most cited references9
- Record: found
- Abstract: found
- Article: not found
Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: a prospective cohort study
Matthew Cummings, Matthew Baldwin, Darryl Abrams … (2020)
- Record: found
- Abstract: found
- Article: not found
Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China
Hua Su, Ming Ming Yang, Cheng Wan … (2020)