- Record: found
- Abstract: found
- Article: found
Metabarcoding Is Powerful yet Still Blind: A Comparative Analysis of Morphological and Molecular Surveys of Seagrass Communities
Read this article at
Abstract
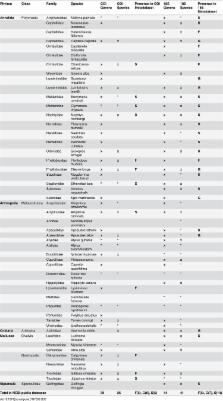
In the context of the sixth wave of extinction, reliable surveys of biodiversity are increasingly needed to infer the cause and consequences of species and community declines, identify early warning indicators of tipping points, and provide reliable impact assessments before engaging in activities with potential environmental hazards. DNA metabarcoding has emerged as having potential to provide speedy assessment of community structure from environmental samples. Here we tested the reliability of metabarcoding by comparing morphological and molecular inventories of invertebrate communities associated with seagrasses through estimates of alpha and beta diversity, as well as the identification of the most abundant taxa. Sediment samples were collected from six Zostera marina seagrass meadows across Brittany, France. Metabarcoding surveys were performed using both mitochondrial (Cytochrome Oxidase I) and nuclear (small subunit 18S ribosomal RNA) markers, and compared to morphological inventories compiled by a long-term benthic monitoring network. A sampling strategy was defined to enhance performance and accuracy of results by preventing the dominance of larger animals, boosting statistical support through replicates, and using two genes to compensate for taxonomic biases. Molecular barcodes proved powerful by revealing a remarkable level of diversity that vastly exceeded the morphological survey, while both surveys identified congruent differentiation of the meadows. However, despite the addition of individual barcodes of common species into taxonomic reference databases, the retrieval of only 36% of these species suggest that the remaining were either not present in the molecular samples or not detected by the molecular screening. This finding exemplifies the necessity of comprehensive and well-curated taxonomic reference libraries and multi-gene surveys. Overall, results offer methodological guidelines and support for metabarcoding as a powerful and repeatable method of characterizing communities, while also presenting suggestions for improvement, including implementation of pilot studies prior to performing full “blind” metabarcoding assessments to optimize sampling and amplification protocols.
Related collections
Most cited references19
- Record: found
- Abstract: found
- Article: not found
The future of biodiversity.

- Record: found
- Abstract: found
- Article: found
Detection of a Diverse Marine Fish Fauna Using Environmental DNA from Seawater Samples
- Record: found
- Abstract: found
- Article: not found