- Record: found
- Abstract: found
- Article: found
ABACAS: algorithm-based automatic contiguation of assembled sequences
Read this article at
Abstract
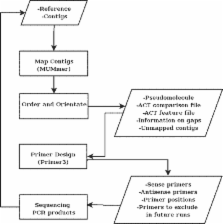
Summary: Due to the availability of new sequencing technologies, we are now increasingly interested in sequencing closely related strains of existing finished genomes. Recently a number of de novo and mapping-based assemblers have been developed to produce high quality draft genomes from new sequencing technology reads. New tools are necessary to take contigs from a draft assembly through to a fully contiguated genome sequence. ABACAS is intended as a tool to rapidly contiguate (align, order, orientate), visualize and design primers to close gaps on shotgun assembled contigs based on a reference sequence. The input to ABACAS is a set of contigs which will be aligned to the reference genome, ordered and orientated, visualized in the ACT comparative browser, and optimal primer sequences are automatically generated.
Availability and Implementation: ABACAS is implemented in Perl and is freely available for download from http://abacas.sourceforge.net
Contact: sa4@ 123456sanger.ac.uk
Related collections
Most cited references9
- Record: found
- Abstract: found
- Article: not found
The impact of next-generation sequencing technology on genetics.

- Record: found
- Abstract: found
- Article: found