- Record: found
- Abstract: found
- Article: found
SDM—a server for predicting effects of mutations on protein stability and malfunction
Read this article at
Abstract
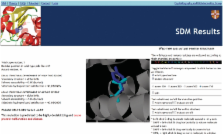
The sheer volume of non-synonymous single nucleotide polymorphisms that have been generated in recent years from projects such as the Human Genome Project, the HapMap Project and Genome-Wide Association Studies means that it is not possible to characterize all mutations experimentally on the gene products, i.e. elucidate the effects of mutations on protein structure and function. However, automatic methods that can predict the effects of mutations will allow a reduced set of mutations to be studied. Site Directed Mutator (SDM) is a statistical potential energy function that uses environment-specific amino-acid substitution frequencies within homologous protein families to calculate a stability score, which is analogous to the free energy difference between the wild-type and mutant protein. Here, we present a web server for SDM ( http://www-cryst.bioc.cam.ac.uk/~sdm/sdm.php), which has obtained more than 10 000 submissions since being online in April 2008. To run SDM, users must upload a wild-type structure and the position and amino acid type of the mutation. The results returned include information about the local structural environment of the wild-type and mutant residues, a stability score prediction and prediction of disease association. Additionally, the wild-type and mutant structures are displayed in a Jmol applet with the relevant residues highlighted.
Related collections
Most cited references61
- Record: found
- Abstract: found
- Article: not found
The complete genome of an individual by massively parallel DNA sequencing.
- Record: found
- Abstract: found
- Article: not found
Very fast empirical prediction and rationalization of protein pKa values.
- Record: found
- Abstract: not found
- Article: not found