- Record: found
- Abstract: found
- Article: found
Chromatin Sampling—An Emerging Perspective on Targeting Polycomb Repressor Proteins
research-article
Robert J. Klose
1
,
* ,
Sarah Cooper
2 ,
Anca M. Farcas
1 ,
Neil P. Blackledge
1 ,
Neil Brockdorff
2
,
*
22 August 2013
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Overview
Polycomb group (PcG) repressor proteins play a central role in gene regulation through
differentiation and development, conferring repressive chromatin configurations at
target gene promoters through their inherent histone modification activities. Recruitment
of Polycomb repressor proteins to defined targets has been attributed to instructive
mechanisms in which sequence-specific binding proteins and/or noncoding RNAs interact
biochemically with the major Polycomb repressive complexes and thus define their sites
of action. Here we highlight that this viewpoint is increasingly incompatible with
experimental observations. We propose an alternative perspective based on the concept
that Polycomb recruitment is responsive rather than instructive. Specifically, we
suggest that Polycomb complexes sample permissive chromatin sites, and through positive
feedback mechanisms, accumulate at those sites lacking antagonistic chromatin modifying
activities linked to ongoing transcription.
Background
PcG repressor proteins were first discovered in Drosophila, where they play a specific
role in maintaining the normal segmental patterns of Hox gene expression through successive
cell generations. Conversely, Trithorax group (TrxG) factors were identified based
on their capacity to maintain the expression of Hox gene loci. Subsequent studies
revealed that both PcG and TrxG proteins are highly conserved in multicellular organisms,
where they perform an essential and pervasive role in epigenetic regulation of gene
expression in differentiation and development [1]. Early studies focused on the capacity
of PcG factors to establish stable heritable silencing at target gene promoters [2],[3].
However, more recent genome-wide studies have revealed that PcG silencing is more
dynamic than previously appreciated [4]. This is most apparent in mammalian cells
where the identity of PcG factor–associated gene promoters often varies significantly
between specific cell types [5]–[7]. Similarly, during X chromosome inactivation in
mammals, recruitment of PcG factors is reversible, being dependent on continuous expression
of the ncRNA Xist [8].
Biochemical and genetic studies have revealed that PcG proteins generally associate
with one of two multi-subunit chromatin modifying complexes, called Polycomb repressive
complex (PRC) 1 and 2 (reviewed in [9]) (Figure 1A). These complexes catalyze defined
histone tail modifications, with PRC1 mono-ubiquitylating histone H2A (H2AK119ub1)
and PRC2 methylating histone H3 lysine 27 (H3K27me3) (Figure 1A). In vertebrates,
PRC1-related complexes subdivide into canonical forms in which the catalytic subunits
are associated with a homologue of the Drosophila proteins Polycomb (CBX2/4/6/7/8)
and Polyhomeotic (PH1/2/3), and noncanonical forms in which catalytic subunits interact
with one of two closely related proteins RYBP or YAF2 (Figure 1 and [10], [11]). Canonical
PRC1 complexes mediate crosstalk with PRC2 complexes via interaction of a chromodomain
in the CBX proteins with PRC2-mediated H3K27me3 [12], [13].
10.1371/journal.pgen.1003717.g001
Figure 1
Core components of the major PcG complexes in vertebrate cells overlap with CpG islands.
(A) PRC1 complexes catalyze monoubiquitylation of histone H2A lysine 119 (H2AK119ub1).
The catalytic subunits are RING1A/B and one of six PCGF protein homologues, PCGF1–6.
PRC1 exists in canonical and noncanonical forms. Canonical PRC1 complexes comprise
RING1A/B, PCGF2/4, the CBX protein subunit that has a chromodomain (CD) that binds
specifically to PRC2-mediated H3K27me3, and the PH subunit. In noncanonical PRC1,
CBX proteins are substituted by the RYBP/YAF2 subunit and the PH subunit is absent.
Association of additional subunits with noncanonical PRC1 occurs in a manner dependent
on the associated PCGF protein. Thus, PCGF1 complexes, discussed extensively herein,
include the additional subunits BCOR and KDM2B. KDM2B has a zinc finger CxxC domain
(CXXC) that mediates binding to unmethylated CpG dinucleotides. PRC2 complexes methylate
histone H3 lysine 27 (H3K27me1/2/3) and comprise the catalytic EZH2 protein and the
core subunits EED, SUZ12, and RBAP48. JARID2 is a substoichiometric subunit that has
been implicated in PRC2 targeting. (B) PcG complexes occupy CpG islands at target
gene promoters: an example from genome-wide ChIP-seq analysis of mouse embryonic stem
cells illustrating a PcG target gene, Lhx4. The Bio-CAP procedure provides a molecular
readout of the density of unmethylated CpG dinucleotides with peaks corresponding
to CpG islands. ChIP-seq for PRC1 (RING1B) and PRC2 (EZH2) subunits illustrates that
PcG protein occupancy closely mirrors unmethylated CpG density.
TrxG proteins similarly form multiprotein chromatin modifying complexes [14]. These
include the SWI-SNF and NURF histone remodelling complexes and two separate histone
methyltransferase complexes that deposit H3 lysine 4 (H3K4me3) or H3 lysine 36 (H3K36me2/3).
H3K4me3 is deposited at active gene promoters and H3K36me3 over the body of transcribed
loci. The catalytic activities of PcG and TrxG complexes underpin their effector functions
by affecting directly, or indirectly, engagement of the transcriptional machinery.
Instructive Models for PcG Complex Targeting
The recruitment of TrxG complexes has been linked to binding of sequence-specific
transcription factors (TFs) and establishment of transcription complexes. Similarly,
studies examining the Drosophila Hox loci have identified promoter-linked elements,
PREs (polycomb response elements), that function as landing platforms for sequence-specific
binding factors that are thought to directly recruit PcG complexes [3], [15]–[17].
Building on this, it has been proposed that PcG recruitment to Hox loci in Drosophila
occurs in a stepwise process with binding of the sequence-specific TFs, notably Pho,
resulting in direct recruitment of PRC2 complexes. In turn, H3K27me3 deposited on
histones by PRC2 is thought to result in the hierarchical recruitment of canonical
PRC1 through its intrinsic capacity to recognize this modification [18]. This model
has been extrapolated to vertebrate systems where sequence-specific binding factors,
for example YY1, the direct homologue of Pho [19], REST [20]–[22], and Runx1 [23],
have all been suggested to recruit PcG complexes via direct biochemical interactions.
However, these individual examples of transcription factor–specific targeting fail
to account for most PcG occupied sites in vivo, with genome-wide analysis instead
indicating that PcG protein occupancy correlates most precisely with broad domains
delineated by the CpG islands of target genes [24] (Figure 1B). Furthermore, sequence-specific
targeting via TFs is insufficient to account for localization of PcG proteins along
the entire length of the inactive X chromosome (Xi) [25]. Xi targeting of PcG proteins
has instead been attributed to direct interaction of PRC2 with the noncoding RNA (ncRNA)
Xist [26], and several studies have since suggested a wider role for ncRNAs in PcG
recruitment. At this time, the precise molecular mechanisms that underpin these proposed
targeting mechanisms remain poorly defined (reviewed in [27]).
Current models invoking PcG recruitment through direct biochemical interaction with
sequence-specific TFs and ncRNAs are summarized in Figure 2A. A central challenge
with the current conceptual framework is to explain how PcG complexes are capable
of interacting with the diversity of TFs and ncRNAs that would be necessary to account
for the dynamic and lineage-specific PcG localization patterns that occur through
development. This issue has been further exacerbated by the recent observation in
mammals that noncanonical PRC1 complexes occupy their normal target gene promoters
in different cell types and localize to the inactive X chromosome independently of
PRC2 and H3K27me3, albeit at reduced levels [10]. As such, for TFs and ncRNA to fulfil
the role as direct recruitment factors it would be necessary for PRC2 and variant
PRC1 complexes to have independently evolved the capability to bind to the same diverse
complement of targeting molecules.
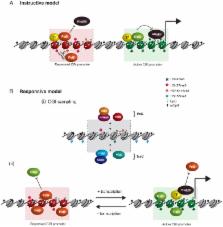
10.1371/journal.pgen.1003717.g002
Figure 2
Comparison of instructive and responsive models for recruitment of PcG complexes to
CpG islands at target gene promoters.
DNA methylation and hypomethylation are indicated with filled and open lollipops,
respectively. Repressed PcG–associated CpG islands are colored red and active TrxG–associated
CpG islands green. (A) Classical instructive models invoke that sequence-specific
DNA binding transcription factors (TF) and/or long noncoding RNAs (lncRNA) interact
biochemically with PcG complexes and thereby target these complexes to defined promoters
at which PcG-mediated histone modifications inhibit RNA polymerase II (RNAPII) activity.
At active gene promoters (large arrow), TFs directly recruit TrxG proteins that in
turn deposit histone modifications linked to gene activation. (B) In the responsive
model, it is proposed that both PcG (PRC1-KDM2B and PRC2) and TrxG (CFP-SET1 and MLL)
complexes stochastically sample unmethylated CpG island chromatin irrespective of
the transcriptional state of a given gene (i). This occurs either via CxxC zinc finger
protein binding to unmethylated CpG or, in the case of PRC2, via sensing the absence
of otherwise pervasive histone modifications, like H3K36me2. The outcome of PcG and
TrxG sampling would then be responsive to the transcriptional state of the associated
gene (ii). In the absence of transcription, the PcG protein–occupied chromatin state
would accumulate by default (aided by positive feedback loops) and antagonize TrxG
activity. Conversely, at transcribed genes, TFs, RNAPolII, and transcription would
favor accumulation of TrxG factors with their associated activities, in turn antagonizing
the function of PcG complexes. Importantly, in the responsive model, the capacity
of PcG proteins to sample CpG island chromatin would permit them to respond to the
transcriptional state of potential target genes without a requirement for direct interactions
with TFs or ncRNAs.
A Responsive Model for Targeting PcG Complexes
In light of accumulating challenges to the concept of instructive targeting of PcG
complexes, we propose an alternative model based not on direct recruitment by sequence-specific
TFs and ncRNAs, but rather on the recognition by PcG complexes of common chromatin
features at target loci. This simplified and generic targeting regime would permit
the concerted establishment of PcG repression at a subset of inactive loci, while
counteracting features elsewhere would limit PcG protein activity. Within the confines
of this conceptual framework, we envisage PcG activity at target loci will respond
to the transcriptional state of the associated gene, as opposed to the prevailing
view which posits that PcG protein function instructs transcriptional outcomes via
physical interaction with and targeting by sequence-specific binding factors.
Key features of our model, focusing on PcG recruitment at target gene promoters, are
illustrated in Figure 2B. The model is guided by the observation that PcG occupancy
in vertebrate cells maps precisely to unmethylated CpG islands (CGI) at target gene
promoters and the contention that these may function as the mammalian equivalent to
PRE [24]. CGIs are short (1 to 2 kilobase) contiguous regions of DNA that escape the
pervasive CpG dinucleotide DNA methylation characteristic of vertebrate genomes. Outside
of CpG islands, DNA methylation represses transcription and contributes to repressive
chromatin states. In contrast, CGIs are generally permissive to transcription and
found in chromatin that is considerably more accessible than surrounding regions of
methylated DNA [28]–[30]. Recent studies suggest that this relies, at least in part,
on the activity of a family of zinc finger-CxxC (ZF-CxxC) DNA binding domain–containing
proteins which recognize nonmethylated CpG dinucleotides and recruit chromatin modifying
activities [31], [32]. CGIs are associated with more than half of vertebrate gene
promoters, of which a subset in any given tissue appear to be very specifically occupied
by the PcG proteins [33]. In fitting with a role for CGIs in PcG protein occupancy,
the extent of PcG protein binding at individual loci correlates precisely with unmethylated
CpG density within the CGI, not with the gene promoter or TSS [24]. Furthermore, an
elegant series of genome engineering studies suggest that CGI characteristics are
sufficient to recruit PcG proteins to ectopic sites [34], [35]; and related to this,
a number of studies have identified a reciprocal relationship between PcG occupancy
and DNA methylation [36]–[38].
In support of a role for CGIs in PcG recruitment, a direct molecular link between
unmethylated CGIs and PcG localization has recently emerged. KDM2B, a ubiquitously
expressed ZF-CxxC domain–containing protein, forms a noncanonical PRC1 complex which
can bind to nonmethylated CpG sites [39]–[41]. Recognition of nonmethylated DNA by
KDM2B allows this variant PRC1 complex to associate with most CGIs, albeit at levels
which are extremely low in comparison to the levels of PRC1 that are observed at the
subset of PcG-repressed CGI targets [41]. We propose that dynamic association of KDM2B
with CGIs provides a means for PRC1 to sample potential target sites. Accumulation
of PRC1 at a limited number of sites could then occur as a consequence of PRC1 monitoring
the chromatin/transcription state within this limited search space. This simple but
elegant sampling module would afford the necessary flexibility to allow PRC1 to engage
all potential target sites in the genome, but ultimately only establish high-level
PcG protein occupancy and silencing in a given cell type at a subset of susceptible
CGIs. Importantly, this model circumvents the necessity for direct targeting of PRC1
by cell type–specific TFs, yet still allows a disparate complement of CGI target genes
to be selected in developmentally diverse lineages. Moreover, KDM2B-mediated PRC1
sampling would be restricted to CGIs as CpG dinucleotides are underrepresented elsewhere
in the genome and are pervasively methylated.
Could a similar CGI sampling activity also play a role in localization of PRC2 complexes?
At present, there is no evidence for PRC2 binding preferentially to unmethylated CpG
dinucleotides, but Jarid2, a substoichiometric PRC2 component, has been reported to
bind GC-rich DNA sequences [42], . Alternatively, PRC2 selectivity for certain CGI
targets might simply be achieved by its inherent preference for specific chromatin
configurations and the limited capacity of its methyltransferase activity to modify
histone tails with certain cis modifications [44], [45]. In support of this possibility,
PRC2 activity is drastically inhibited by TrxG-mediated H3K4me3, a modification highly
enriched at actively transcribed genes. Interestingly, the mammalian TrxG H3K4me3
methyltransferase complexes include proteins that contain ZF-CxxC domains, suggesting
a dynamic CGI sampling mechanism could also guide TrxG and H3K4me3 to CGI promoters.
PRC2 activity is also inhibited by a second modification, H3K36me2/me3. H3K6me2 is
notable in this context as it is an abundant modification found at high levels throughout
the genome but depleted at CGIs as a consequence of H3K36me1/2 demethylase activity
of the ZF-CxxC domain proteins KDM2A and KDM2B [32], [46]. Thus, cis-inhibitory effects
of these, and possibly other histone H3 tail modifications, may serve to define a
chromatin state which is permissive to PRC2 occupancy and modification.
The idea that a dynamic sampling process may underpin the functionality of PcG complexes
on chromatin is supported by the observation that PRC2 redistributes in mammalian
cell lines [36]–[38], [47], [48] and in higher plant cells [49], [50] when DNA methylation
is depleted. Similarly, in germline cells of C. elegans embryos, mutations in the
MES4 protein, the major methyltransferase catalyzing H3K36me2, result in redistribution
of PRC2 from the X chromosome where it is normally enriched to sites throughout the
autosomes [51]. In both of these examples, loss of chromatin modifications that normally
function to inhibit PcG activity in bulk chromatin appears to allow PcG activity to
migrate to new sites, effectively titrating it away from bona fide PcG target sites.
Feedback Mechanisms Stabilizing PcG and TrxG Chromatin States
If CGI hypomethylation, and linked to this, CGI-specific chromatin configurations,
define sites where variant PRC1 and PRC2 can engage productively, what are the mechanisms
that restrict H3K27me3 and H2AK119ub1 accumulation to nonexpressed targets? As outlined
above, we propose that in a given cell type the subset of CGI sites that ultimately
become PcG targets transition from a “sampled” state into an “established” PcG protein
occupied and repressed state through the concerted action of both PRC1 and PRC2. This
may be mediated through positive feedback loops driven by the function of their chromatin
modifying activities. For example, PRC2-mediated H3K27me3 can lead to hierarchical
recruitment of canonical PRC1 complexes via CBX proteins which bind H3K27me3. Furthermore,
recently it was shown that PRC2 prefers to methylate compact chromatin substrates
[44], a feature that PRC1 may potentiate [9].
Conversely, the majority of CGIs that remain free of established PcG silencing must
possess a degree of “anti-silencing” that counteracts PcG protein establishment. How
might this be achieved? One simple explanation would be that the major activity underpinning
anti-silencing at most CGI promoters is the occupancy of positively acting transcription
factors, the transcriptional machinery itself, and the function of TrxG proteins at
these sites. Based on our evolving molecular understanding of PcG protein activity,
there may be some sense in this idea. For example, although there exists some inherent
level of H3K4me3 at CpG islands through ZF-CxxC domain–mediated targeting mechanisms
[31], productive transcription dramatically amplifies this through RNA polymerase
II–dependent recruitment of additional H3K4 methyltransferase activity [52]–[54].
As indicated above, PRC2-mediated H3K27 methylation is directly inhibited when the
histone H3 tail carries H3K4me3. During activated gene expression, CGI regions also
become extensively histone acetylated on H3 at position 27 (H3K27ac) which could have
the added effect of directly blocking PRC2 activity and H3K27me3 [55]. Fittingly,
it was recently shown that PRC2 fails to methylate H3K27 at some established PcG sites
in cells lacking the NURD deacetylase complex which deacetylates H3K27ac [56]. Interestingly,
several mammalian H3K4 methyltransferase complexes also associate with H3K27 demethylases,
indicating a concerted effort to counteract PcG activity at CGIs during the process
of H3K4me3 deposition and transcription [57].
If PcG proteins simply function to identify CGI loci that already lack significant
transcriptional activity, in a manner that is responsive to transcription, why would
their silencing services be required at all? One possibility lies in the fact that
PcG systems appear to play important roles in keeping genes off in tissues where they
should not normally be expressed. During development, as a cell transitions from one
lineage to another, the expression of a subset of genes which are specific to the
parental cell type must diminish in expression. A constant regime of CGI sampling
would provide an opportunity for PcG proteins to identify these CGI sites in the genome,
presumably by their lack of activated transcription and reduced capacity to anti-silence.
Full and productive engagement of both PRC1 and PRC2 at these sites could then initiate,
amplify, and fully establish a classical polycomb-repressed domain in response to
this diminished anti-silencing. This would allow the cell to partition genes that
have lost the capacity to be efficiently transcribed, an outcome that would be beneficial
as it would help to protect the transcriptional identity of the cell from stochastic
gene expression events that could lead to aberrant functionality. Furthermore, it
would force the cell to invoke strong and persistent transcriptional signals to activate
genes necessary for the transition to new and alternative transcriptional states.
Not inconsistent with these ideas, removal of PcG proteins in embryonic stem cells
affects transcription levels of only a proportion of target loci and then only by
a small increment [24], [58]–[60]. We would suggest that this limited activation occurs
as a result of basal activating signals present within the cell that are not of sufficient
magnitude to overcome normal PcG-mediated silencing barriers.
The model we propose for PcG protein function in vertebrates is mechanistically rooted
in an underlying requirement for nonmethylated DNA at CGIs, a feature that is not
conserved in many invertebrate species which generally lack pervasive genome DNA methylation.
Nevertheless, we believe that conceptually the interplay between “silencing” and “anti-silencing”
at CGIs very much parallels the observations and ideas about PcG protein function
that have emerged through the genetic and molecular study of the PcG system in Drosophila.
For example, the type of positively acting chromatin modifying activities (remodelling
ATPases, H3K4 and H3K36 methyltransferases) that we suggest are amplified with transcription
and mechanistically correspond to anti-silencing at CGIs were originally identified
in genetic interaction screens in Drosophila as PcG-counteracting TrxG proteins [14].
The direct mechanisms that lead to PcG and TrxG protein targeting to PREs in Drosophila
still remain incompletely described. However, in light of our model, it is tempting
to speculate that alternative sampling processes which do not rely on nonmethylated
DNA may be at play in selecting PcG protein occupied sites in Drosophila. Indeed,
there is indirect evidence that supports the possibility that PREs are continually
sampled for susceptibility to PcG establishment [4], [61]. Moreover, the historical
view that these outcomes should rely on TF-mediated interactions are not inconsistent
with our model, but instead we would suggest these relationships may be less direct
in nature with the system responding to the activity of TFs at PREs as opposed to
directly mediating their outcomes.
Broader Implications for the Role of Chromatin Structure in Transcriptional Regulation
The concept of CGIs as sampling platforms for chromatin modifying enzymes has broader
implications for how we view chromatin structure as part of transcription. If as we
propose PcG and TrxG proteins respond, as opposed to instruct transcription, these
factors would likely act as important modulators of transcriptional outcomes as opposed
to drivers. This may be achieved by the creation of a bistable chromatin switch at
CGIs through the opposing activities of PcG and TrxG complexes. For example, if PcG
proteins can sample CGIs and then identify those that are in the nontranscribed state,
positive feedback mechanisms linking PRC1 and PRC2 could help to reinforce a monostable
chromatin state that is inhibitory to transcription (Figure 3). If CGIs are constantly
sampled by both PcG and TrxG chromatin modifying activities, an opportunity to switch
the chromatin state may constantly exist. For example, targeting of a strong and concerted
activating signal to a PcG-occupied CGI promoter followed by productive transcription
could permit exit from the PcG protein–repressed chromatin state coupled with transcription-associated
deposition of H3K4me3. Several mammalian TrxG proteins encode domains that recognize
H3K4me3, including the H3K4 methyltransferases themselves, perhaps leading to a transcription
and TrxG-based positive feedback loop that defines a second transcriptionally permissive
monostable chromatin state. Given that TrxG complexes in mammals contain activities
that directly oppose PcG protein repressive function, this active chromatin state
may play a modulatory role in sustaining transcription. Following cessation of transcription,
the TrxG reinforcement and positive feedback loop sustained by this activity may correspondingly
diminish. This transcription/TrxG anti-silencing effect could eventually be insufficient
to counteract the PcG inhibitory state which may be reacquired by default. The net
effect of this type of modulatory system presumably would be to ensure that the appropriate
type and strength of activating signal is present before a gene is turned on and that
the transcribed state can be sustained as long as the appropriate activating signal
is present. Furthermore, individual chromatin states maintained by feedback mechanisms
could contribute to mitotic heritability as defined in classical genetic experiments
on PcG and TrxG systems.
10.1371/journal.pgen.1003717.g003
Figure 3
A simplified representation of the responsive model emphasizing that it has properties
of a classical bistable switch.
A schematic of the proposed bistable switch (center panel). The ground state at CpG
island chromatin, PcG occupancy (left panel), is the default that is overcome only
when TF-mediated gene activation reaches a critical threshold. Positive feedback mechanisms
between PRC1 and PRC2 reinforce PcG occupancy and antagonize the TrxG state. Beyond
the activation threshold, the system switches to the TrxG state (right panel) that
is in turn reinforced by positive feedback mechanisms between RNAPII and TrxG factors.
This, together with TrxG-mediated antagonism of PcG activities, ensures stability
of the TrxG state such that stochastic fluctuations in gene activation signal would
not trigger a switch back to the PcG state. However, when gene activation signals
drop below a critical threshold, the CpG island will switch back to the default PcG
state.
Interestingly, high-resolution mapping of histone modifications in mammalian cells
has revealed a class of repressed PcG CGI promoters at which TrxG-mediated H3K4me3,
histone acetylation, and RNA polymerase II occupancy are all detectable, albeit at
relatively low levels, referred to as bivalent promoters [62]–[64]. In the context
of our model, we interpret that bivalency could represent the outcome of stochastic
sampling of PcG-established CGIs by TrxG ZF-CxxC proteins or a degree of activator
signal which is insufficient to switch the chromatin state. It will be interesting
to examine in more detail the phenomena of bivalency in the context of a potential
bistable chromatin state.
In support of the concept of chromatin bistability at CGIs, recent studies examining
PcG chromatin modifications, TrxG chromatin modifications, and gene expression during
stem cell differentiation also propose a bistable chromatin state that appears to
describe the system with some degree of accuracy [65], [66]. Importantly, these studies
suggest that local CpG density contributed by CGIs would be an important feature of
such a system. Furthermore, modelling of chromatin dynamics and transcription during
the process of plant vernalization, which utilizes polycomb systems, has also proposed
that polycomb functions as part of a bistable chromatin switch [67]. Clearly, it will
be interesting within the context of these concepts to design kinetic experiments
to test the validity of such models in mammals and formally examine whether chromatin
modulates transcriptional outputs consistent with the properties attributed to bistable
systems.
Concluding Comments
In summary, we suggest that our responsive model for PcG protein function in vertebrates
overcomes many of the issues that complicate the prevailing instructive models, notably
the requirement to invoke that PcG complexes physically interact with a unique set
of TFs or ncRNAs in individual cell types. It should however be noted that the two
types of model are not necessarily mutually exclusive. Furthermore, many of the features
of our model adhere to the general principles describing PcG and TrxG protein function
that were developed around observations from early genetic studies which suggest that
these systems do not define transcriptional states but instead function to maintain
predetermined transcriptional states [68]. In this respect, we believe the core molecular
attributes of our model will provide a useful conceptual framework on which to experimentally
examine its predictions and begin to better understand how the PcG and TrxG proteins
function in gene regulation. Furthermore, it highlights the possibility that CGIs
as core components of most vertebrate gene promoters may play a central role in modulating
gene expression by providing a gene regulatory platform that is capable of contributing
to a bistable chromatin environment at gene promoters. Although our description deals
primarily with PcG recruitment at target gene CGIs in mammalian cells, we consider
that the mechanisms invoked may be universal and at some level contribute to PcG recruitment
at sites such as Xi, and also in other model systems, for example C. elegans, higher
plants, and Drosophila.
Note Added in Proof
A review of bivalent gene promoters that reaches similar conclusions to some of those
expressed in this viewpoint was published when this article was under revision [69].
Acknowledgments
Formulation of our model required the synthesis of many ideas and observations suggested
previously by others and we wish to unequivocally acknowledge the derivative nature
of our proposal. Due to limitations in the scope and depth of our viewpoint, we may
have failed to reference all relevant papers and we apologize for any instances where
this has occurred. We wish to thank Tom Milne, our lab colleagues, and the wider community
for stimulating discussions.
Related collections
Most cited references47
- Record: found
- Abstract: found
- Article: not found
A chromatin landmark and transcription initiation at most promoters in human cells.
Matthew Guenther, Stuart S. Levine, Laurie A. Boyer … (2007)
- Record: found
- Abstract: found
- Article: not found
Chromatin signatures of pluripotent cell lines.
Véronique Azuara, Pascale Perry, Stephan Sauer … (2006)
- Record: found
- Abstract: found
- Article: not found
Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors.
Fabio Mohn, Michael Weber, Michael Rebhan … (2008)