- Record: found
- Abstract: found
- Article: found
Western Bluetongue virus serotype 3 in Sardinia, diagnosis and characterization
Read this article at
Abstract
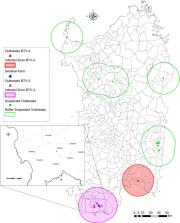
Over the last 20 years, Italy has experienced multiple incursions of different serotypes of Bluetongue virus ( BTV), a Culicoides‐borne arbovirus, the causative agent of bluetongue ( BT), a major disease of ruminants. The majority of these incursions originated from Northern Africa, likely because of wind‐blown dissemination of infected midges. Here, we report the first identification of BTV‐3 in Sardinia, Italy. BTV‐3 circulation was evidenced in sentinel animals located in the province of Sud Sardegna on September 19, 2018. Prototype strain BTV‐3 SAR2018 was isolated on cell culture. BTV‐3 SAR2018 sequence and partial sequences obtained by next‐generation sequencing from nucleic acids purified from the isolate and blood samples, respectively, were demonstrated to be almost identical (99–100% of nucleotide identity) to BTV‐3 TUN2016 identified in Tunisia in 2016 and 2017, a scenario already observed in past incursions of other BTV serotypes originating from Northern Africa.
Related collections
Most cited references13
- Record: found
- Abstract: found
- Article: not found
Cloning of a human parvovirus by molecular screening of respiratory tract samples.
- Record: found
- Abstract: found
- Article: not found
Sequence analysis of bluetongue virus serotype 8 from the Netherlands 2006 and comparison to other European strains.

- Record: found
- Abstract: found
- Article: found