- Record: found
- Abstract: found
- Article: found
Prader-Willi Syndrome and Growth Hormone Deficiency
Read this article at
Abstract
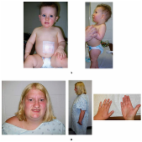
Prader-Willi syndrome (PWS) is a rare multisystem genetic disorder demonstrating great variability with changing clinical features during patient’s life. It is characterized by severe hypotonia with poor sucking and feeding difficulties in early infancy, followed by excessive eating and gradual development of morbid obesity in later infancy or early childhood. The phenotype is most probably due to hypothalamic dysfunction which is also responsible for growth hormone (GH) and thyroid-stimulating hormone (TSH) deficiencies, central adrenal insufficiency and hypogonadism. The multidimensional problems of patients with PWS can be managed with multidisciplinary approach. Reduced GH secretion, low peak GH response to stimulation, decreased spontaneous GH secretion and low serum IGF-1 levels in PWS patients have been documented in many studies. GH therapy has multiple beneficial effects on growth and body composition, motor and mental development in PWS patients. The recommended dosage for GH is 0.5-1 mg/m2/day. GH therapy should not be started in the presence of obstructive sleep apnea syndrome, adenotonsillar hypertrophy, severe obesity and diabetes mellitus. GH treatment should be considered for patients with genetically confirmed PWS in conjunction with dietary, environmental and life-style measures.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: found
Prader-Willi Syndrome: Obesity due to Genomic Imprinting
- Record: found
- Abstract: found
- Article: not found
Clinical report—health supervision for children with Prader-Willi syndrome.
- Record: found
- Abstract: found
- Article: found