- Record: found
- Abstract: found
- Article: found
Widespread Compensatory Evolution Conserves DNA-Encoded Nucleosome Organization in Yeast
Read this article at
Abstract
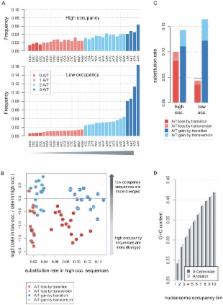
Evolution maintains organismal fitness by preserving genomic information. This is widely assumed to involve conservation of specific genomic loci among species. Many genomic encodings are now recognized to integrate small contributions from multiple genomic positions into quantitative dispersed codes, but the evolutionary dynamics of such codes are still poorly understood. Here we show that in yeast, sequences that quantitatively affect nucleosome occupancy evolve under compensatory dynamics that maintain heterogeneous levels of A+T content through spatially coupled A/T-losing and A/T-gaining substitutions. Evolutionary modeling combined with data on yeast polymorphisms supports the idea that these substitution dynamics are a consequence of weak selection. This shows that compensatory evolution, so far believed to affect specific groups of epistatically linked loci like paired RNA bases, is a widespread phenomenon in the yeast genome, affecting the majority of intergenic sequences in it. The model thus derived suggests that compensation is inevitable when evolution conserves quantitative and dispersed genomic functions.
Author Summary
Purifying selection is a major force in conserving genomic features. It pushes deleterious mutations to extinction while conserving the specific DNA sequence. Here we show that a large proportion of the yeast genome evolves under compensatory dynamics that conserve genomic properties while modifying the genomic sequence. Such compensatory evolution conserves the local G+C content of the genome, which influences nucleosome organization. Since purifying selection is too weak to eliminate every weakly deleterious mutation in nucleosome bound or unbound sequences, the local G+C content is frequently stabilized by compensatory G+C gaining and G+C losing mutations in proximal loci. Theoretical analysis shows that compensatory evolution is inevitable when natural selection is weak and the genomic feature is distributed over many loci. These results imply that sequence conservation may not always be equated with overall selection. They demonstrate that cycles of weakly deleterious substitutions followed by positive selection for corrective mutations, which were so far studied mostly in RNA coding genes, are observed broadly and profoundly affect genome evolution.
Related collections
Most cited references38
- Record: found
- Abstract: found
- Article: not found
ChIP-seq accurately predicts tissue-specific activity of enhancers.
- Record: found
- Abstract: found
- Article: not found
Population genomics of domestic and wild yeasts
- Record: found
- Abstract: found
- Article: not found