- Record: found
- Abstract: found
- Article: found
Long-term Eculizumab Therapy in a Child With Refractory Immune Complex–Mediated Membranoproliferative Glomerulonephritis
case-report

Rahul Chanchlani
1
,
2
,
5 ,
Paul Thorner
3
,
4 ,
Seetha Radhakrishnan
1
,
4 ,
Diane Hebert
1
,
4 ,
Valerie Langlois
1
,
4 ,
Steven Arora
5 ,
David Barth
6
,
7 ,
Daniel Cattran
6
,
7 ,
Michael Kirschfink
8 ,
Christoph Licht
1
,
4
,
9
,
∗
08 September 2017
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Introduction
There have been recent developments in the understanding of the pathogenesis of membranoproliferative
glomerulonephritis (MPGN) supporting a prominent role for the complement alternative
pathway (AP).1, 2 MPGN due to AP dysregulation has been further classified into dense-deposit
disease (DDD) and C3 glomerulonephritis (C3GN), and grouped together as C3 glomerulopathy
(C3G). This entity includes all glomerular lesions that are characterized by predominant
C3 accumulation with minimum or scant Ig deposition and highlights the pathogenetic
contribution of complement.1, 2 On the other hand, MPGN secondary to autoimmune diseases
or infections is labeled as immune complex−mediated MPGN.
C3G is associated with a poor prognosis, as 30% to 50% of patients progress to end-stage
renal disease (ESRD) within 10 years of diagnosis, and around 50% have recurrence
after transplantation.
1
Complement targeting therapy such as eculizumab has recently emerged as a novel therapeutic
option for patients with C3G. There have been few case reports to describe the effectiveness
of eculizumab in patients with C3G, but the literature is scarce in the pediatric
population.3, 4, 5 Moreover, there is very little insight into the long-term safety
and efficacy regarding the use of eculizumab in immune complex−mediated MPGN. We present
a child with refractory immune complex−mediated MPGN who was successfully treated
with eculizumab for a period of 4 years.
Case Report
A 16-year-old girl presented with a 2-month history of edema, anemia, hypertension,
microscopic hematuria, nephrotic range proteinuria, low C3 level, and was diagnosed
as immune complex−mediated MPGN on renal biopsy (Figure 1). Treatment with prednisone
and mycophenolate mofetil (MMF) was started (Table 1). Her detailed clinical picture
has been published earlier.
4
Briefly, her proteinuria persisted and 4 months later, she was admitted with fever,
pancytopenia, seizures, and pneumonia secondary to Pseudomonas aeruginosa sepsis.
Her renal function worsened, and hemodialysis and plasmapheresis were initiated along
with appropriate supportive treatment.
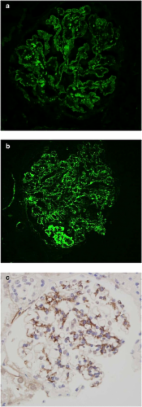
Figure 1
Histopathological features before receiving eculizumab. Biopsy findings pre-eculizumab.
(a) Biopsy samples show a membranoproliferative appearance by light microscopy on
periodic acid–Schiff staining. There is strong IgG staining (b) and less intense C3
staining (c). (d) By electron microscopy, there are subendothelial deposits (black
arrow) and mesangial interposition (white arrow) with effacement of podocyte foot
processes (*). Findings establish a diagnosis of immune complex−mediated membranoproliferative
glomerulonephritis. (a−c, original magnification ×400; d, original magnification ×10,000.)
Table 1
Baseline clinical and laboratory characteristics of the child with refractory membranoproliferative
glomerulonephritis
Baseline characteristics
Results
Age at onset, yr
16
Presenting features
Edema, anemia, hypertension, microscopic hematuria and nephrotic range proteinuria
Hemoglobin (normal, 12–15 g/dl)
7.5
Platelets (normal, 150–400 × 103/μl)
688
Creatinine (normal, 0.6–1.02 mg/dl)
0.9
Albumin (normal, 3–5 g/dl)
1.7
C3 (normal, 83–152 mg/dl)
15
C4 (normal, 13–37 mg/dl)
13
Urine protein to creatinine ratio (normal, < 0.2 mg/mg)
4.8
Immunosuppression received before eculizumab
Prednisone and MMF
Complement analysis
CFH, CFHR5, CFI, CFB
No mutation
C3
No mutation
MCP, TBHD
No mutation
CFHR1
Present
C3NeF
Present
sC5b-9 levels (normal, < 320 ng/ml)
844
Anti−factor H antibody
Absent
CFB, complement factor B; CFH, complement factor H; CFHR-1: complement factor H related
protein 1; CFHR-5, complement factor H related protein 5; CFI, complement factor I;
C3NeF, C3 nephritic factor; MCP, membrane cofactor protein; NA, not available; THBD,
thrombomodulin.
Conversion factor: serum creatinine in mg/dl to μl/l: × 88.4.
Interestingly, complement analysis revealed low C3, high soluble membrane attack complex
(sMAC, sC5b-9) level (844 ng/ml; normal < 320 ng/ml), low CH50, positive C3NeF, and
absent CFHR 1 on western blot, interpreted as strong evidence for complement AP activation,
possibly driven by a CFHR 1 deficiency and positive C3NeF. She received 9 sessions
of plasmapheresis over a period of 11 days before commencing eculizumab. Initially,
she received 4 doses of eculizumab (900 mg/wk for 4 weeks) followed by a rapid (i.e.,
within days) improvement in neurological complications and hematological parameters.
Hemodialysis was stopped after 2 weeks. After the sixth dose, her proteinuria had
significantly improved (urine protein to creatinine ratio 0.04 mg/mg from a baseline
of 3.89 mg/mg). Thereafter, she was maintained on biweekly eculizumab infusions of
1200 mg each. She did not have any side effects related to eculizumab. Prednisone
was weaned and stopped over a period of 1 year after starting eculizumab. Figure 2
shows variation in the laboratory parameters from 3 months before until 48 months
after initiating eculizumab.
Figure 2
Changes in laboratory parameters before and after initiating eculizumab. The laboratory
values are shown from 3 months before until 48 months after starting eculizumab. For
conversion to SI units: (a) albumin (g/l) = g/dl × 10; creatinine (μl/l) = mg/dl ×
88.4; C3 (g/l) = mg/dl × 0.01. HD, hemodialysis; PLEX, plasma exchange; UPUC, urine
protein to urine creatinine ratio.
Four years later, she continues to be on maintenance eculizumab (1200 mg every 2 weeks)
without other immunosuppressive or antihypertensive medications. She has non−nephrotic-range
proteinuria (urine protein-to-creatinine ratio at 51 months after eculizumab, 0.39
mg/mg). Her estimated glomerular filtration rate (eGFR) is 96.5 ml/min per 1.73 m2,
but she continues to have positive C3NeF (normal: negative), low C3, elevated C3d
(86 mU/l; normal < 40 mU/l), and elevated sMAC levels (606 ng/ml; normal < 320 ng/ml).
Discussion
MPGN is a rare, yet an important cause of glomerulopathy affecting children and young
adults. The pathogenesis of MPGN may either be immune complex mediated or attributed
to the defect in complement AP regulation, including mutations in the genes coding
for C3
6
or complement inhibitors such as complement factor H (CFH), complement factor I (CFI),
or membrane cofactor protein (MCP; CD46),
7
or the presence of C3NeF.7, 8 In this report, the patient was considered to be C3G
based on obvious AP abnormalities, but her renal biopsy showed dominant staining of
both Igs and C3. Acknowledging the strict histopathological criteria for C3G (i.e.,
dominant C3 staining ≤ 2 intensity levels above Ig staining), we continued to label
her condition as immune complex−mediated MPGN.
There are no evidence-based guidelines for treatment in patients with C3G. Conventional
therapies such as angiotensin-converting enzyme inhibitors, immunosuppressive agents
and plasma exchange/infusions have been used but with variable results.9, 10, 11 As
activation of the complement AP is thought to be a major patho-mechanism in C3G, the
use of complement-targeting and complement-control−restoring therapies such as eculizumab
have evolved as an obvious treatment option (Table 2).
Table 2
Teaching points
1. MPGN is a rare, yet an important cause of glomerulopathy affecting children and
young adults.
2. MPGN due to alternate complement pathway dysregulation has been further classified
into DDD and C3GN, and grouped together as C3G.
3. There are no evidence-based guidelines for treatment in patients with C3G.
4. Eculizumab is a humanized anti-C5 monoclonal antibody, and its role in treatment
of C3G has been emerging.
5. Few patients with immune complex−mediated MPGN may have abnormalities in alternate
complement pathway, and they may benefit from the use of eculizumab.
C3G, C3 glomerulopathy; C3GN, C3 glomerulonephritis; DDD, dense-deposit disease; MPGN,
membranoproliferative glomerulonephritis.
Eculizumab is a humanized anti-C5 monoclonal antibody that has been successfully used
in patients with atypical hemolytic uremic syndrome. There is emerging evidence for
the role of eculizumab in the treatment of C3G. There have been few case reports3,
4, 5, 12, 13 and case series14, 15 describing the effect of eculizumab in C3G, however,
with mixed results.
The literature is particularly limited with respect to the long-term implications
of the use of eculizumab in children having immune complex−mediated MPGN. We present
the long-term clinical profile of a young patient with immune complex−mediated MPGN
who was treated with eculizumab for 4 years after conventional therapy had failed
to control the disease. She became dialysis independent, and proteinuria improved
after initiation of eculizumab. The infusions were well tolerated, with no major side
effects. Our report adds to the current understanding of this relatively new medication
in this rare disease.
Importantly, the patient continued to have significantly low C3 levels even after
4 years of therapy, suggesting that, unlike in atypical hemolytic uremic syndrome,
effective complement inhibition in MPGN may also need inhibition of C3 convertase.
This finding is in keeping with the concept of MPGN being disease driven by unrestricted
C3 activation in fluid phase vs. atypical hemolytic uremic syndrome being caused by
unrestricted activation of the terminal complement pathway on the level of the vascular
endothelium.
16
Not all patients with C3G are likely to benefit from eculizumab. It seems useful in
those with relatively short disease duration, active inflammatory renal lesions (extensive
endocapillary proliferation and crescents), limited glomerular and interstitial fibrosis
on a (recent) kidney biopsy sample, and increased circulating serum sC5b-9 levels.
17
Our study is limited, as it was restricted to 1 patient only. Also, a repeat renal
biopsy after eculizumab therapy to document changes in the histopathology after therapy
was not available. Nevertheless, our report demonstrates that a few patients with
histopathological features of immune complex−mediated MPGN who are resistant to conventional
treatment may have abnormalities in the alternate complement pathway and may benefit
from the use of complement targeting therapy. It will be worthwhile to explore whether
all cases of immune complex−mediated MPGN without clear underlying etiology should
undergo workup for abnormalities in the alternate complement pathway.
In conclusion, eculizumab appears to be a safe and effective therapeutic option in
pediatric patients with immune complex−mediated MPGN. Further prospective studies
in a larger patient cohort will be required to better understand the long-term clinical
implications of eculizumab treatment in pediatric patients with immune complex−mediated
MPGN, with a special emphasis on determining the optimal duration of treatment.
Disclosure
CL serves on advisory boards of Alexion Pharmaceuticals, Inc. and has received travel
and speaker stipends as well as unrestricted research grants from Alexion Pharmaceuticals,
Inc. MK has received fees from Alexion Pharmaceuticals, Inc. for invited lectures.
All the other authors declared no conflict of interest.
The patient and her family provided written informed consent to this publication.
Related collections
Most cited references15
- Record: found
- Abstract: found
- Article: not found
Eculizumab for dense deposit disease and C3 glomerulonephritis.
E C Heher, Yuzhou Zhang, M. Stokes … (2012)