- Record: found
- Abstract: found
- Article: found
Copy number variation is associated with gene expression change in archaea
Abstract
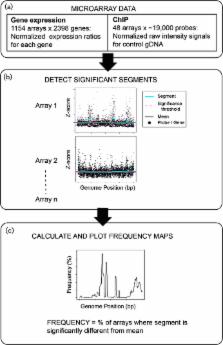
Genomic instability, although frequently deleterious, is also an important mechanism for microbial adaptation to environmental change. Although widely studied in bacteria, in archaea the effect of genomic instability on organism phenotypes and fitness remains unclear. Here we use DNA segmentation methods to detect and quantify genome-wide copy number variation (CNV) in large compendia of high-throughput datasets in a model archaeal species, Halobacterium salinarum. CNV hotspots were identified throughout the genome. Some hotspots were strongly associated with changes in gene expression, suggesting a mechanism for phenotypic innovation. In contrast, CNV hotspots in other genomic loci left expression unchanged, suggesting buffering of certain phenotypes. The correspondence of CNVs with gene expression was validated with strain- and condition-matched transcriptomics and DNA quantification experiments at specific loci. Significant correlation of CNV hotspot locations with the positions of known insertion sequence (IS) elements suggested a mechanism for generating genomic instability. Given the efficient recombination capabilities in H. salinarum despite stability at the single nucleotide level, these results suggest that genomic plasticity mediated by IS element activity can provide a source of phenotypic innovation in extreme environments.
Related collections
Most cited references53
- Record: found
- Abstract: not found
- Article: not found
Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing
- Record: found
- Abstract: found
- Article: not found
Global variation in copy number in the human genome.
- Record: found
- Abstract: found
- Article: not found