- Record: found
- Abstract: found
- Article: found
LATS2 overexpression attenuates the therapeutic resistance of liver cancer HepG2 cells to sorafenib-mediated death via inhibiting the AMPK–Mfn2 signaling pathway
Read this article at
Abstract
Background
Effective therapy for hepatocellular carcinoma (HCC) is currently an imperative issue, and sorafenib is a first-line drug for the treatment of HCC. However, the clinical benefit of sorafenib is often impaired by drug resistance. Accordingly, the present study was conducted to investigate the molecular mechanisms involving sorafenib resistance, with a focus on large tumor suppressor 2 (LATS2) and mitophagy.
Methods
HepG2 liver cancer cells were treated with sorafenib and infected with adenovirus-loaded LATS2 (Ad-LATS2). Cell death, proliferation and migration were measured via western blotting analysis, immunofluorescence and qPCR. Mitochondrial function and mitophagy were determined via western blotting and immunofluorescence.
Results
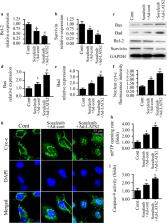
Our data indicated that LATS2 expression was repressed by sorafenib treatment, and overexpression of LATS2 could further enhance sorafenib-mediated apoptosis in HepG2 liver cancer cells. At the molecular level, mitochondrial stress was triggered by sorafenib treatment, as evidenced by decreased mitochondrial membrane potential, increased mitochondrial ROS production, more cyc-c release into the nucleus, and elevated mitochondrial pro-apoptotic proteins. However, in response to mitochondrial damage, mitophagy was activated by sorafenib treatment, whereas LATS2 overexpression effectively inhibited mitophagy activity and thus augmented sorafenib-mediated mitochondrial stress. Subsequently, we also demonstrated that the AMPK–MFN2 signaling pathway was involved in mitophagy regulation after exposure to sorafenib treatment and/or LATS2 overexpression. Inhibition of the AMPK pathway interrupted mitophagy and thus enhanced the antitumor property of sorafenib, similar to the results obtained via overexpression of LATS2.
Related collections
Most cited references59

- Record: found
- Abstract: found
- Article: found
Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy

- Record: found
- Abstract: found
- Article: found
Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission‐VDAC1‐HK2‐mPTP‐mitophagy axis
- Record: found
- Abstract: found
- Article: not found