- Record: found
- Abstract: found
- Article: found
Long-read RNA sequencing of human and animal filarial parasites improves gene models and discovers operons
Read this article at
Abstract
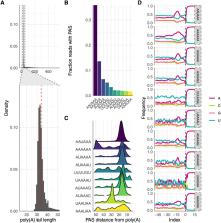
Filarial parasitic nematodes (Filarioidea) cause substantial disease burden to humans and animals around the world. Recently there has been a coordinated global effort to generate, annotate, and curate genomic data from nematode species of medical and veterinary importance. This has resulted in two chromosome-level assemblies ( Brugia malayi and Onchocerca volvulus) and 11 additional draft genomes from Filarioidea. These reference assemblies facilitate comparative genomics to explore basic helminth biology and prioritize new drug and vaccine targets. While the continual improvement of genome contiguity and completeness advances these goals, experimental functional annotation of genes is often hindered by poor gene models. Short-read RNA sequencing data and expressed sequence tags, in cooperation with ab initio prediction algorithms, are employed for gene prediction, but these can result in missing clade-specific genes, fragmented models, imperfect mapping of gene ends, and lack of isoform resolution. Long-read RNA sequencing can overcome these drawbacks and greatly improve gene model quality. Here, we present Iso-Seq data for B. malayi and Dirofilaria immitis, etiological agents of lymphatic filariasis and canine heartworm disease, respectively. These data cover approximately half of the known coding genomes and substantially improve gene models by extending untranslated regions, cataloging novel splice junctions from novel isoforms, and correcting mispredicted junctions. Furthermore, we validated computationally predicted operons, manually curated new operons, and merged fragmented gene models. We carried out analyses of poly(A) tails in both species, leading to the identification of non-canonical poly(A) signals. Finally, we prioritized and assessed known and putative anthelmintic targets, correcting or validating gene models for molecular cloning and target-based anthelmintic screening efforts. Overall, these data significantly improve the catalog of gene models for two important parasites, and they demonstrate how long-read RNA sequencing should be prioritized for ongoing improvement of parasitic nematode genome assemblies.
Author summary
Filarial parasitic nematodes are vector-borne parasites that infect humans and animals. Brugia malayi and Dirofilaria immitis are transmitted by mosquitoes and cause human lymphatic filariasis and canine heartworm disease, respectively. Recent years have seen a dramatic increase in genomic and transcriptomic data sets and the concomitant increase in innovative strategies for drug target identification, validation, and screening. However, while the completeness of genome assemblies of filarial parasitic nematodes has seen steady improvements, the reliability of gene models has not kept pace, hindering cloning efforts. Long-read RNA sequencing technologies are uniquely able to improve gene models, but have not been widely used for the causative agents of neglected tropical diseases. Here, we report the improvement of gene models in both B. malayi and D. immitis by long-read RNA sequencing. We identified novel operons, deprecated false positive operons, identified dozens of novel genes, and described the parameters of polyadenylation. We also focused on putative anthelmintic targets, identifying novel isoforms and correcting gene models. These data substantially increase the trustworthiness of gene models in these two species and demonstrate how long-read sequencing approaches should be prioritized in the continued improvement of genome assemblies and their gene annotations.
Related collections
Most cited references67

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools

- Record: found
- Abstract: found
- Article: found
BEDTools: a flexible suite of utilities for comparing genomic features

- Record: found
- Abstract: found
- Article: found