- Record: found
- Abstract: found
- Article: found
Diversity analysis of MSP1 identifies conserved epitope organization in block 2 amidst high sequence variability in Indian Plasmodium falciparum isolates

Read this article at
Abstract
Background
Despite its immunogenicity, the polymorphic nature of merozoite surface protein 1, an important vaccine candidate for Plasmodium falciparum malaria, remains a concern. This study analyses the impact of genetic variability and parasite population structure on epitope organization of different MSP1 segments.
Methods
Altogether 98 blood samples collected from P. falciparum infected mild and severe malaria patients of Chhattisgarh and West Bengal were used to sequence regions encoding block 2 and MSP1-19 of msp1. Sequences were analysed using MEGA7, DnaSPv5, Arlequin3.5 and BepiPred.
Results
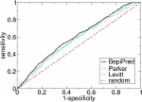
All three major MSP1 block 2 allele families namely K1, MAD20 and RO33 were detected in the samples and they together resulted in 41 indel variants. Chhattisgarh samples displayed an average MOI of 2.07 ± 1.59 which was higher in mild malaria and in age group < 18 years. Ultra-structure of block 2 alleles revealed that mutation and repeat expansion were two major mechanisms responsible for allelic variability of K1 and MAD20. Regions flanking block 2 were highly variable in Chhattisgarh with average mismatch differences (k) ranging from 1.198 to 5.156 for three families. In contrast, region encompassing MSP1-19 exhibited limited heterogeneity (k Chhattisgarh = 1.45, k West Bengal = 1.363). Of the 50 possible B cell linear epitopes predicted from block 2 variants, 94.9% (131 of 138) of the parasites could be represented by three conserved antigens.
Conclusions
Present data indicates that natural selection and transmission intensity jointly play a role in controlling allelic diversity of MSP1 in Indian parasite isolates. Despite remarkable genetic variability, a limited number of predominant and conserved epitopes are present in Indian parasite isolates reinstating the importance of MSP1 as a promising malaria vaccine candidate.
Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: found
Arlequin (version 3.0): An integrated software package for population genetics data analysis
- Record: found
- Abstract: found
- Article: found