- Record: found
- Abstract: found
- Article: found
Jab1 is a target of EGFR signaling in ERα-negative breast cancer
Read this article at
Abstract
Introduction
c-Jun activation domain-binding protein-1 (Jab1) is a multifunctional signaling protein that previously has been shown to be a master regulator of a poor prognostic gene signature in invasive breast cancer and to mediate the action of S100A7. Since epidermal growth factor receptor (EGFR), like S100A7, is often expressed in estrogen receptor-alpha-negative (ERα -) breast cancer, we set out to investigate the role of Jab1 in mediating EGFR signaling, another facet of the ERα - phenotype.
Methods
MDA-MB-231 and MDA-MB-468 ERα -/EGFR + cell lines were assessed for localization of Jab1 and levels of downstream genes by immunofluorescence and nuclear protein extract assay following treatment with epidermal growth factor (EGF) and extracellular signal-regulated kinase (ERK) pathway inhibitor. A cohort of 424 human breast tumors was also assessed by immunohistochemistry.
Results
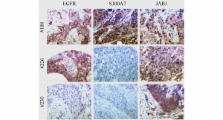
EGF treatment of cell lines resulted in increased Jab1 nuclear expression. This effect was inhibited by the ERK pathway inhibitor, PD98059. EGF treatment was also associated with colocalization of pERK (phosphorylated ERK) and Jab1 as well as regulation of the Jab1 downstream target gene, p27. When Jab1 activity was knocked down, p27 levels were restored to pre-EGF treatment level. Analysis of EGFR and Jab1 expression in a cohort of invasive breast tumors by tissue microarray and immunohistochemistry confirmed a relationship between EGFR and increased nuclear Jab1 within the ERα - subset (n = 154, P = 0.019). The same association was also confirmed for S100A7 and Jab1 ( P = 0.036), and high Jab1 nuclear expression was most frequent in tumors that were positive for both EGFR and S100A7 ( P = 0.004).
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest.
- Record: found
- Abstract: found
- Article: not found
NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis.
- Record: found
- Abstract: found
- Article: not found