- Record: found
- Abstract: found
- Article: found
Mitochondrial function as a therapeutic target in heart failure
Read this article at
Abstract
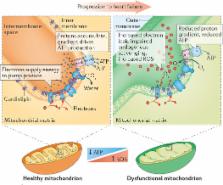
Heart failure is a pressing worldwide public-health problem with millions of patients having worsening heart failure. Despite all the available therapies, the condition carries a very poor prognosis. Existing therapies provide symptomatic and clinical benefit, but do not fully address molecular abnormalities that occur in cardiomyocytes. This shortcoming is particularly important given that most patients with heart failure have viable dysfunctional myocardium, in which an improvement or normalization of function might be possible. Although the pathophysiology of heart failure is complex, mitochondrial dysfunction seems to be an important target for therapy to improve cardiac function directly. Mitochondrial abnormalities include impaired mitochondrial electron transport chain activity, increased formation of reactive oxygen species, shifted metabolic substrate utilization, aberrant mitochondrial dynamics, and altered ion homeostasis. In this Consensus Statement, insights into the mechanisms of mitochondrial dysfunction in heart failure are presented, along with an overview of emerging treatments with the potential to improve the function of the failing heart by targeting mitochondria.
Related collections
Most cited references214
- Record: found
- Abstract: found
- Article: not found
Antioxidants prevent health-promoting effects of physical exercise in humans.
- Record: found
- Abstract: found
- Article: not found
Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury.
- Record: found
- Abstract: not found
- Article: not found