- Record: found
- Abstract: found
- Article: found
Using Linkage Analysis to Detect Gene-Gene Interactions. 2. Improved Reliability and Extension to More-Complex Models
Read this article at
Abstract
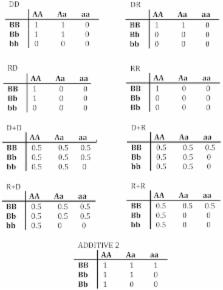
Detecting gene-gene interaction in complex diseases has become an important priority for common disease genetics, but most current approaches to detecting interaction start with disease-marker associations. These approaches are based on population allele frequency correlations, not genetic inheritance, and therefore cannot exploit the rich information about inheritance contained within families. They are also hampered by issues of rigorous phenotype definition, multiple test correction, and allelic and locus heterogeneity. We recently developed, tested, and published a powerful gene-gene interaction detection strategy based on conditioning family data on a known disease-causing allele or a disease-associated marker allele 4. We successfully applied the method to disease data and used computer simulation to exhaustively test the method for some epistatic models. We knew that the statistic we developed to indicate interaction was less reliable when applied to more-complex interaction models. Here, we improve the statistic and expand the testing procedure. We computer-simulated multipoint linkage data for a disease caused by two interacting loci. We examined epistatic as well as additive models and compared them with heterogeneity models. In all our models, the at-risk genotypes are “major” in the sense that among affected individuals, a substantial proportion has a disease-related genotype. One of the loci ( A) has a known disease-related allele (as would have been determined from a previous analysis). We removed (pruned) family members who did not carry this allele; the resultant dataset is referred to as “stratified.” This elimination step has the effect of raising the “penetrance” and detectability at the second locus ( B). We used the lod scores for the stratified and unstratified data sets to calculate a statistic that either indicated the presence of interaction or indicated that no interaction was detectable. We show that the new method is robust and reliable for a wide range of parameters. Our statistic performs well both with the epistatic models (false negative rates, i.e., failing to detect interaction, ranging from 0 to 2.5%) and with the heterogeneity models (false positive rates, i.e., falsely detecting interaction, ≤1%). It works well with the additive model except when allele frequencies at the two loci differ widely. We explore those features of the additive model that make detecting interaction more difficult. All testing of this method suggests that it provides a reliable approach to detecting gene-gene interaction.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Parametric and nonparametric linkage analysis: a unified multipoint approach.
- Record: found
- Abstract: found
- Article: not found
The power to detect linkage in complex disease by means of simple LOD-score analyses.
- Record: found
- Abstract: found
- Article: not found