- Record: found
- Abstract: found
- Article: found
Paternal leakage and mtDNA heteroplasmy in Rhipicephalus spp. ticks
Read this article at
Abstract
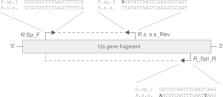
Paternal leakage of mitochondrial DNA (mtDNA) and heteroplasmy have been recently described in several animal species. In arthropods, by searching in the Scopus database, we found only 23 documented cases of paternal leakage. Therefore, although arthropods represent a large fraction of animal biodiversity, this phenomenon has been investigated only in a paucity of species in this phylum, thus preventing a reliable estimate of its frequency. Here, we investigated the occurrence of paternal leakage and mtDNA heteroplasmy in ticks belonging to one of the most significant tick species complexes, the so-called Rhipicephalus sanguineus sensu lato. By developing a multiplex allele-specific PCR assay targeting a fragment of the 12S rRNA ribosomal region of the mtDNA, we showed the occurrence of paternal leakage and mtDNA heteroplasmy in R. sanguineus s.l. ticks originated from experimental crosses, as well as in individuals collected from the field. Our results add a new evidence of paternal leakage in arthropods and document for the first time this phenomenon in ticks. Furthermore, they suggest the importance of using allele-specific assays when searching for paternal leakage and/or heteroplasmy, as standard sequencing methods may fail to detect the rare mtDNA molecules.
Related collections
Most cited references51
- Record: found
- Abstract: found
- Article: not found
Bias in template-to-product ratios in multitemplate PCR.
- Record: found
- Abstract: found
- Article: not found