- Record: found
- Abstract: found
- Article: not found
Tumor cells convert immature myeloid dendritic cells into TGF-β–secreting cells inducing CD4 + CD25 + regulatory T cell proliferation
Read this article at

Abstract
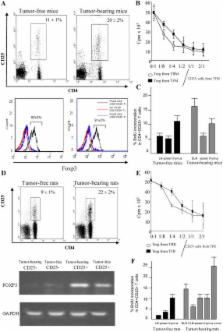
The mechanisms through which regulatory T cells accumulate in lymphoid organs of tumor-bearing hosts remain elusive. Our experiments indicate that the accumulation of CD4 + CD25 + regulatory T cells (T reg cells) expressing FoxP3 and exhibiting immunosuppressive function originates from the proliferation of naturally occurring CD25 + T cells and requires signaling through transforming growth factor (TGF)– β receptor II. During tumor progression, a subset of dendritic cells (DCs) exhibiting a myeloid immature phenotype is recruited to draining lymph nodes. This DC subset selectively promotes the proliferation of T reg cells in a TGF- β–dependent manner in mice and rats. Tumor cells are necessary and sufficient to convert DCs into regulatory cells that secrete bioactive TGF- β and stimulate T reg cell proliferation. In conclusion, tumor expansion can stimulate T reg cells via a specific DC subset.
Related collections
Most cited references24
- Record: found
- Abstract: found
- Article: not found
Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state.
- Record: found
- Abstract: found
- Article: not found
Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells.
- Record: found
- Abstract: found
- Article: not found