- Record: found
- Abstract: found
- Article: found
Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders
Abstract
Background
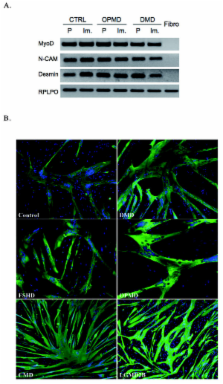
Investigations into both the pathophysiology and therapeutic targets in muscle dystrophies have been hampered by the limited proliferative capacity of human myoblasts. Isolation of reliable and stable immortalized cell lines from patient biopsies is a powerful tool for investigating pathological mechanisms, including those associated with muscle aging, and for developing innovative gene-based, cell-based or pharmacological biotherapies.
Methods
Using transduction with both telomerase-expressing and cyclin-dependent kinase 4-expressing vectors, we were able to generate a battery of immortalized human muscle stem-cell lines from patients with various neuromuscular disorders.
Results
The immortalized human cell lines from patients with Duchenne muscular dystrophy, facioscapulohumeral muscular dystrophy, oculopharyngeal muscular dystrophy, congenital muscular dystrophy, and limb-girdle muscular dystrophy type 2B had greatly increased proliferative capacity, and maintained their potential to differentiate both in vitro and in vivo after transplantation into regenerating muscle of immunodeficient mice.
Conclusions
Dystrophic cellular models are required as a supplement to animal models to assess cellular mechanisms, such as signaling defects, or to perform high-throughput screening for therapeutic molecules. These investigations have been conducted for many years on cells derived from animals, and would greatly benefit from having human cell models with prolonged proliferative capacity. Furthermore, the possibility to assess in vivo the regenerative capacity of these cells extends their potential use. The innovative cellular tools derived from several different neuromuscular diseases as described in this report will allow investigation of the pathophysiology of these disorders and assessment of new therapeutic strategies.
Related collections
Most cited references35
- Record: found
- Abstract: found
- Article: not found
Extension of life-span by introduction of telomerase into normal human cells.
- Record: found
- Abstract: found
- Article: not found
Telomere measurement by quantitative PCR.

- Record: found
- Abstract: found
- Article: found