- Record: found
- Abstract: found
- Article: found
Epitope-Based Immunoinformatics and Molecular Docking Studies of Nucleocapsid Protein and Ovarian Tumor Domain of Crimean–Congo Hemorrhagic Fever Virus
Read this article at
Abstract
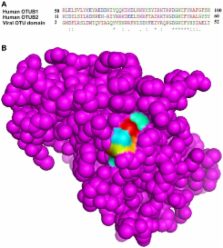
Crimean–Congo hemorrhagic fever virus (CCHFV), the fatal human pathogen is transmitted to humans by tick bite, or exposure to infected blood or tissues of infected livestock. The CCHFV genome consists of three RNA segments namely, S, M, and L. The unusual large viral L protein has an ovarian tumor (OTU) protease domain located in the N terminus. It is likely that the protein may be autoproteolytically cleaved to generate the active virus L polymerase with additional functions. Identification of the epitope regions of the virus is important for the diagnosis, phylogeny studies, and drug discovery. Early diagnosis and treatment of CCHF infection is critical to the survival of patients and the control of the disease. In this study, we undertook different in silico approaches using molecular docking and immunoinformatics tools to predict epitopes which can be helpful for vaccine designing. Small molecule ligands against OTU domain and protein–protein interaction between a viral and a host protein have been studied using docking tools.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: not found
Structure validation by Calpha geometry: phi,psi and Cbeta deviation.
- Record: found
- Abstract: found
- Article: not found
A semi-empirical method for prediction of antigenic determinants on protein antigens.
- Record: found
- Abstract: found
- Article: found