- Record: found
- Abstract: found
- Article: found
Genomic Insights into the Atopic Eczema-Associated Skin Commensal Yeast Malassezia sympodialis
Read this article at
ABSTRACT
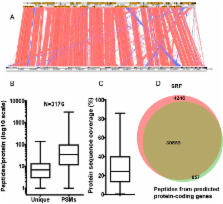
Malassezia commensal yeasts are associated with a number of skin disorders, such as atopic eczema/dermatitis and dandruff, and they also can cause systemic infections. Here we describe the 7.67-Mbp genome of Malassezia sympodialis, a species associated with atopic eczema, and contrast its genome repertoire with that of Malassezia globosa, associated with dandruff, as well as those of other closely related fungi. Ninety percent of the predicted M. sympodialis protein coding genes were experimentally verified by mass spectrometry at the protein level. We identified a relatively limited number of genes related to lipid biosynthesis, and both species lack the fatty acid synthase gene, in line with the known requirement of these yeasts to assimilate lipids from the host. Malassezia species do not appear to have many cell wall-localized glycosylphosphatidylinositol (GPI) proteins and lack other cell wall proteins previously identified in other fungi. This is surprising given that in other fungi these proteins have been shown to mediate interactions (e.g., adhesion and biofilm formation) with the host. The genome revealed a complex evolutionary history for an allergen of unknown function, Mala s 7, shown to be encoded by a member of an amplified gene family of secreted proteins. Based on genetic and biochemical studies with the basidiomycete human fungal pathogen Cryptococcus neoformans, we characterized the allergen Mala s 6 as the cytoplasmic cyclophilin A. We further present evidence that M. sympodialis may have the capacity to undergo sexual reproduction and present a model for a pseudobipolar mating system that allows limited recombination between two linked MAT loci.
IMPORTANCE
Malassezia commensal yeasts are associated with a number of skin disorders. The previously published genome of M. globosa provided some of the first insights into Malassezia biology and its involvement in dandruff. Here, we present the genome of M. sympodialis, frequently isolated from patients with atopic eczema and healthy individuals. We combined comparative genomics with sequencing and functional characterization of specific genes in a population of clinical isolates and in closely related model systems. Our analyses provide insights into the evolution of allergens related to atopic eczema and the evolutionary trajectory of the machinery for sexual reproduction and meiosis. We hypothesize that M. sympodialis may undergo sexual reproduction, which has important implications for the understanding of the life cycle and virulence potential of this medically important yeast. Our findings provide a foundation for the development of genetic and genomic tools to elucidate host-microbe interactions that occur on the skin and to identify potential therapeutic targets.
Related collections
Most cited references87
- Record: found
- Abstract: found
- Article: not found
Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training.
- Record: found
- Abstract: found
- Article: not found
Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models.
- Record: found
- Abstract: found
- Article: not found