- Record: found
- Abstract: found
- Article: found
Myogenic Akt signaling upregulates the utrophin–glycoprotein complex and promotes sarcolemma stability in muscular dystrophy
Read this article at
Abstract
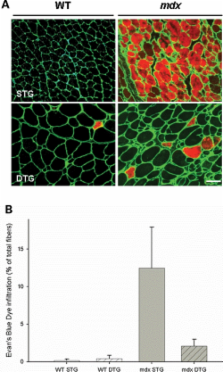
Duchenne muscular dystrophy is caused by dystrophin mutations that lead to structural instability of the sarcolemma membrane, myofiber degeneration/regeneration and progressive muscle wasting. Here we show that myogenic Akt signaling in mouse models of dystrophy promotes increased expression of utrophin, which replaces the function of dystrophin thereby preventing sarcolemma damage and muscle wasting. In contrast to previous suggestions that increased Akt in dystrophy was a secondary consequence of pathology, our findings demonstrate a pivotal role for this signaling pathway such that modulation of Akt can significantly affect disease outcome by amplification of existing, physiological compensatory mechanisms.
Related collections
Most cited references21
- Record: found
- Abstract: found
- Article: not found
The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription.
- Record: found
- Abstract: found
- Article: not found
Expression of full-length utrophin prevents muscular dystrophy in mdx mice.
- Record: found
- Abstract: found
- Article: not found