- Record: found
- Abstract: found
- Article: found
SDM: a server for predicting effects of mutations on protein stability
Read this article at
Abstract
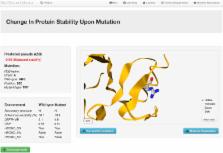
Here, we report a webserver for the improved SDM, used for predicting the effects of mutations on protein stability. As a pioneering knowledge-based approach, SDM has been highlighted as the most appropriate method to use in combination with many other approaches. We have updated the environment-specific amino-acid substitution tables based on the current expanded PDB (a 5-fold increase in information), and introduced new residue-conformation and interaction parameters, including packing density and residue depth. The updated server has been extensively tested using a benchmark containing 2690 point mutations from 132 different protein structures. The revised method correlates well against the hypothetical reverse mutations, better than comparable methods built using machine-learning approaches, highlighting the strength of our knowledge-based approach for identifying stabilising mutations. Given a PDB file (a Protein Data Bank file format containing the 3D coordinates of the protein atoms), and a point mutation, the server calculates the stability difference score between the wildtype and mutant protein. The server is available at http://structure.bioc.cam.ac.uk/sdm2
Related collections
Most cited references52

- Record: found
- Abstract: found
- Article: found
DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach
- Record: found
- Abstract: found
- Article: not found
Prediction of protein stability changes for single-site mutations using support vector machines.
- Record: found
- Abstract: found
- Article: not found