
INTRODUCTION
Influenza and other acute respiratory infections in Russia are still far more common and uncontrolled infections. According to The Russian Ministry of Health data, more than 80% of all infectious diseases in Russia fall to the share of influenza and acute respiratory infections, and their economic impact is approx. 85% of the total damage caused by infectious diseases. Influenza is an example of a disease caused by a virus, a constant evolution of which leads to annual epidemics. The essential feature of influenza viruses’ evolution is associated with the accumulation of mutations leading to antigenic drift and appearance of new viral strains resulting in heterogeneity of viral populations and formation of different genetic lines [1]. The relationship between the dynamic prevalence of certain phylogenetic lines of influenza viruses and their geographical distribution was registered during the past decade. Dramatic changes in the genetic structure of influenza A virus can evolve by the reassortment of the genomic segments of different subtypes circulating in humans and animals [2]. The emergence of a new pandemic strain of influenza virus A(H1N1pdm2009) in 2009 confirmed the potential of the evolutionary variability and unpredictable shift of the influenza virus.
Continuous etiological surveillance of influenza on the national and global level carried out in the framework of cooperation between National Centers for Influenza and Collaborating Centers for Influenza of the World Health Organization (WHO) is of great importance for the investigation of virus evolution and forecast of the next epidemic season. In this paper, we describe the molecular evolution of influenza A and B viruses circulated in Russia in 2006-2013.
The goal of this research project was to study the natural variability of human influenza A and B viruses, based on the analysis of the population structure of influenza viruses circulating in Russia in order to determine the direction of their genetic and antigenic drift by comparison to the WHO reference strains.
MATERIALS AND METHODS
Viruses
In this paper we present the results of analysis of more than 420 strains of influenza A and B viruses isolated from clinical specimens (nasopharyngeal swabs and throat, bronchial-alveolar lavage, cell material) during seven epidemic seasons (2006-2013) in the Research Institute of Influenza as well as received from 33 Regional Basic Laboratories according to “Rospotrebnadzor” program fromthecitiesAstrakhan,Belgorod,Cheboksary,Volgograd, Vologda, Vladivostok, Yekaterinburg, Irkutsk, Kaliningrad, Krasnoyarsk, Kursk, Lipetsk, Moscow, Murmansk, Nizhny Novgorod, Novosibirsk, Orenburg, Omsk, Petropavlovsk-Kamchatsky, Rostov-on-Don, Ryazan, Samara, St. Petersburg, Smolensk, Stavropol, Tomsk, Tver, Tula, Ulan-Ude, Khabarovsk, Chita, Chelyabinsk, and Yakutsk. Reference strains were obtained from the Centers for Disease Control and Prevention (CDC Atlanta,GA,USA).
Virus isolation was performed in MDCK cell culture and 10-day-old chicken embryos (CE) incubated for 72 hours at 34°C according to the approved method [3].
Hemagglutination Inhibition (HI) test
HI test was performed according to the standard method recommended by the WHO, with 0.75% suspension of human red blood cells group 0 for all epidemic viruses. In the case of pandemic influenza A(H1N1)pdm09 isolates 1% suspension of chicken erythrocytes was used [4].
Antisera
Influenza reference antisera obtained from the WHO Reference Center (CDC, Atlanta, USA) and rat strain specific antisera to Russian representative isolates and WHO reference strains were prepared in the Research Institute of Influenza.
Sequencing
RNA extraction was made using commercial RNeasy Mini Kit (Qiagen, Germany). Reverse transcription of the RNA was performed by Reverta-L kit (InterLabService, Russia). The amplification of cDNA was performed by the standard method using the original primers. Sequencing of influenza virus A genome fragments (genes HA, NA, M, NS, and NP) was carried out on ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems, USA) with “BigDye Terminator Cycle Sequencing Kit v3.1”.
Phylogenetic analysis
The processing and analysis of sequences was performed using the software Vector NTI v10.1.1 (Invitrogen) and MEGA 5 (PSU, USA). The method of maximum likelihood (ML) was used to build phylogenetic trees. The choice of evolutionary models was implemented by Akaike criterion (AIC) value using ModelTest v3.7 program [5]. Algorithm SLAC was used to search for sites of positive selection [6].
RESULTS AND DISCUSSION
The results of the analysis of the antigenic properties and genetic structure of representative samples of influenza A and B strains circulated in the Russian Federation in 2006-2012 epidemic seasons are presented below.
Influenza viruses A(H1N1)
Influenza A(H1N1) viruses in Russia during this period, as in other countries, occurred more rarely than influenza A(H3N2) and B viruses.The etiological structure of influenza viruses during 7 epidemic seasons (2006-2013) is shown in Table 1. The percentage of detection of these viruses to the total number of influenza viruses among patients with influenza-like illness or acute respiratory infection (ILI/ ARI) gradually decreased from 42% in 2006-2007 to 19% in 2008-2009. Significant epidemic events caused by this pathogen were recorded infrequently. However, the gradual accumulation of point mutations in HA and NA genes led to changes in the antigenic profile of the surface proteins and the emergence of a new drift variant of A(H1N1) virus.
| Epidemic period | Viruses of each subtype/type as a percentage to the total number of influenza viruses among Russian ILI/ARI patients | |||
|---|---|---|---|---|
| A(H1N1) | A(H1N1)pdm09 | A(H3N2) | B* | |
| 2006-2007 | 42 | 0 | 32 | 26 |
| 2007-2008 | 38 | 0 | 29 | 33 (94% Vic) |
| 2008-2009 | 19 | 0 | 63 | 18 (98% Yam) |
| 2009-2010 | 0 | 99 | 0 | 1 (100% Vic) |
| 2010-2011 | 0 | 65 | 10 | 25 (100% Vic) |
| 2011-2012** | 0 | 1 | 63 | 36 (70% Vic) |
| 2012-2013 | 0 | 41 | 39 | 20 (87% Yam) |
% of prevalent lineage (Yamagata (Yam) or Victoria (Vic)) is indicated in brackets.
The season of very low influenza activity.
All A(H1N1) strains isolated in 2006-2007 could be divided into two antigenically separate groups: a group that differed from both the old (A/New Caledonia/20/1999) and the new (A/Solomon Islands/3/2006) vaccine strains and the group which was very close to the new reference strain A/Solomon Islands/3/2006. However, a population of the latter A(H1N1) viruses was quite small in number and, therefore, a vaccine containing this component, would not be fully protective against the disease. Phylogenetic analysis showed that 2006-2007 influenza A viruses of H1N1 subtype were also related to two genetic groups - clades 1 and 2. Strains belonging to clade 1 were similar to A/New Caledonia/20/1999 vaccine strain (Fig. 1). Viruses containing specific substitutions T82K, Y94H, R145K, R208K, R225K in the hemagglutinin (HA) and V234M, D382N in neuraminidase (NA) formed genetically heterogeneous group of clade 2 viruses. The circulation of influenza A clade 2A viruses similar to the A/Solomon Islands/3/2006 vaccine strain was practically not registered during this period in Russia. Viruses that circulated during this period formed a separate group and were the predecessors of clade 2C viruses, which appeared in 2007 with the substitutions S36N, A189T in HA, and S82K, M188I in NA. A minor incidence of A(H1N1) viruses similar to A/Brisbane/59/07 (clade 2B) has been observed in Russia since 2007.

The tree was constructed using maximum likelihood method, CTR+G+I model, bootstrap 1000 replications. Viruses sequenced in the Research Institute of Influenza are marked in bold. ♦ – vaccine viruses.
Influenza A(H1N1) viruses also actively circulated throughout Russia during the next epidemic season (2007-2008) and were isolated in 13 cities. Antigenic analysis of isolates showed that all of them, regardless of the place of isolation, finally lost antigenic relationship with the old reference strain A/New Caledonia/20/1999; they practically did not react with serum antibodies raised against this reference strain.
Those isolates can be divided into two groups by comparison of their antigenic characteristics with A/Solomon Islands/3/2006 reference virus. The first group interacted with the antiserum within 2-fold of the homologous titer. This group contained only 17.2% of the isolated A(H1N1) viruses. The second group reacted with the serum raised against A/Solomon Islands/3/2006 within a fourfold to eightfold reduction of homologous titer. At the same time, the epidemic 2008 isolates interacted quite well with antiserum against A/Brisbane/59/2007 (within to 1-2-fold of homologous titer).
Thus, the majority of influenza A(H1N1) viruses that circulated in Russia in 2007-2008 differed from the reference strains A/New Caledonia/20/1999 and A/ Solomon Islands/3/2006 and were closer to the reference strain A/Brisbane/59/2007 according to their antigenic characteristics. These viruses were continued to be found in the 2008-2009 season. Strains of two antigenic types: A/ Brisbane/59/2007-like and antigenically related to A/Hong Kong/1870/2008 were circulating during this epidemic season in Russia.
A(H1N1) viruses of the 2008-2009 season made up 18.7% of all isolated influenza viruses in this season. Most viruses of this subtype have been isolated in Siberia and the Far East. Viruses similar to A/Brisbane/59/2007 vaccine strain, which belongs to clade 2B, caused this epidemic flu season in Russia. Prevalence of A(H1N1) viruses of clade 2B was typical not only for Russia but for the world as a whole. Among the investigated strains of viruses isolated in the Far East and Siberia, we also found representatives of clade 2C (A/Vladivostok/83/2008, A/ Khabarovsk/3/2009, A/Novosibirsk/9/2009, etc.), which intensively circulated in the Asia-Pacific region [7].
We have shown that HA of Russian clade 2B representatives is characterized by substitutions D35N, R188K, K145R, and clade 2C representatives – by substitutions S36N, K82R, R145K, R188M, A189T and T193K. The differences in the antigenic structure of the representatives of these two clades could be caused by mutations in the coding regions for antigenic site Ca2 (K145R), Cb (K82R) and Sb (T193K). 2008-2009 strains of clade 2B differed from the previously isolated representatives of this clade by the substitution A189T.
The lastA(H1N1) viruses related to the new evolutionary branches have lost any relationship with the old reference strains and interacted poorly with appropriate antisera. Feedbacks were also absent, i.e. serum antibodies to new antigenic variants of the virus did not neutralize strains of previous years. These data suggest significant genetic differences between the viruses of these clusters. Influenza A(H1N1) viruses are characterized by «silent» variability that manifests in the gradual accumulation of amino acid substitutions in the minor undetectable group of viruses. Such variability is often divergent [8, 9]. The emergence and spread of a few antigenically separate clusters of influenza A(H1N1) viruses in 2006-2007 support this hypothesis. The subsequent epidemic events showed that the strains similar to A/St.Petersburg/96/2007 (antigenic variant of A/Brisbane/59/2007 strain) had the largest epidemic potency and selective advantage.
In the HA sequences of influenza A(H1N1) clade 2B representatives, two codons with selective advantages (codons 186 and 227) have been identified (probability more than 95%). According to X-ray analysis, residue 186 is the part of the receptor-binding site of HA (helix 190), which is presumably involved in hydrogen bonding with sialo-pentasaccharide [10, 11].
Substitutions G249K and T287I were detected in NA of clade 2B strains (data not shown), and substitutions M188I, I267M, L367I in 2C clade strains. Substitutions M188I, G249K, T287I and L367I are located in the so- called phylogenetically relevant areas D, F, H and L, respectively. L region of NA of N1 subtype is known to form antigenic site [12]. Most of NA sequences of N1 clade 2B strains have mutation H275Y that leads to oseltamivir resistance, although these strains do not form a separate phylogenetic group within clade 2B. Among the representatives of clade 2C oseltamivir-resistant strains have not been identified.
No strains with resistance determining mutation S31N to rimantadine in the M2 protein were identified among clade 2B strains in contrast to rimantadine resistant strains of clade 2C.
All studied representatives of clade 2C contained the substitution S207N in the M1 protein typical for influenza A(H5N1) strains belonging to Qinghai group (Qinghai-like, e.g. A/bar-headed goose/Qinghai/12/2005). However, the role of this substitution in M1 protein of human influenza A(H1N1) viruses remains unclear. It is known though, that mutations in the C-terminal domain of M1 protein may occur during virus adaptation to mice and are associated with increased pathogenicity of influenza A viruses [13].
Influenza viruses A(H1N1)pdm09
A(H1N1)pdm09 epidemic in Russia began in the middle of September 2009 and had a monoetiologic character. Influenza A(H1N1)pdm09 viruses also dominated in Russia during the 2010-2011 epidemic season (Table 1).
All isolated viruses were homogeneous in their antigenic properties and reacted with A(H1N1)pdm09 diagnostic antiserum obtained from CDC as well as with antiserum raised against A/California/07/2009 strain within 1-4-fold of the homologous titer.
Population of A(H1N1)pdm09 strains was genetically homogeneous and evolutionarily related to five phylogenetic groups [14] belonging to the world dominant clade 7 with characteristic S203T substitution in HA (Fig. 2). All viruses other than those of clade 1 contained P83S substitution in HA. Interestingly, these strains contained valine residue at position 321, which is located near the cleavage site of the HA molecule. However, in a significant number of Russian isolates in 2009 we observed a reversion V321I in HA. A number of 2009-2011 strains contained substitutions in antigenic site Ca, Sb and Sa. Strains isolated in 2010-2011 were characterized by mutational changes that were the result of virus adaptation to the immunized human population. We did not find any significant changes of the genetic structure of HA, NA, M, NS and NP genes of the analyzed strains [15]. None of these strains had oseltamivir resistance mutation (H275Y), but all strains carried the mutation S31N in M2 leading to rimantadine resistance. Most strains (82%) isolated from the autopsy materials of patients with laboratory confirmed flu contained substitution D222G in the HA receptor-binding site.
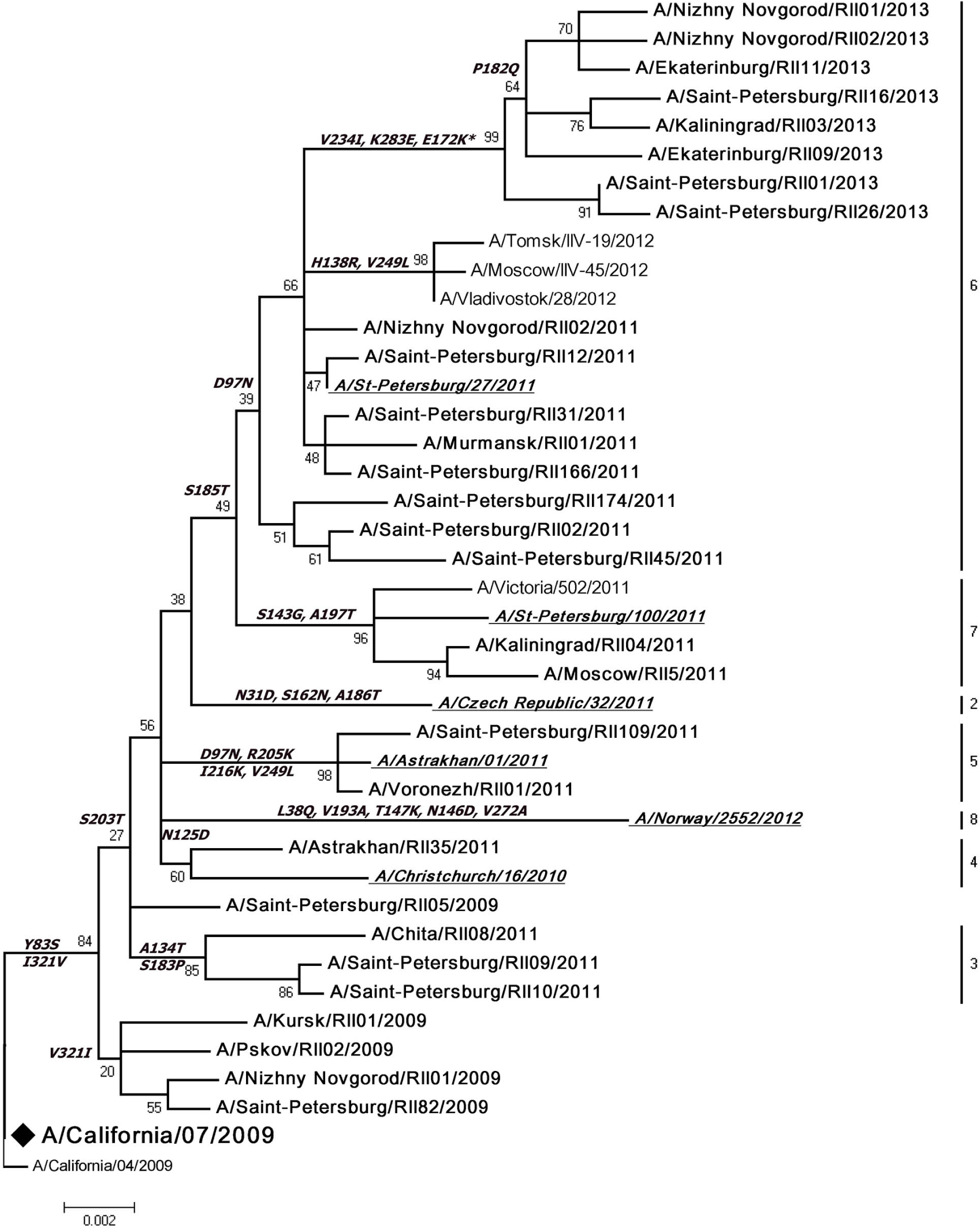
The tree was constructed using maximum likelihood method, TN93+G model, bootstrap 1000 replications. Viruses sequenced in the Research Institute of Influenza are marked in bold. ♦ – vaccine viruses. Genetic groups of reference viruses are shown in italic and underlined.
Our findings confirm that all A(H1N1)pdm09 virus strains of the 2012-2013 epidemic season belonged to clade 6 (A/Saint Petersburg/27/2011-like). Analyzed strains were genetically homogenous contrary to the situation of 2010-2011 season when influenza A(H1N1) pdm09 strains belonged to 5 phylogenetic groups. Sequencing of HA1 subunit of 5 viruses, isolated from autopsy materials, showed amino acid substitutions in four of them: D222G in 2 viruses and Q223R in 2 viruses. No specific antigenic properties were found in influenza A(H1N1)pdm09 viruses isolated from autopsy samples when compared with viruses isolated from patients who recovered from this infection.
The analysis of sites - subjects to positive selection - identified three codons in the HA sequence of influenza A(H1N1)pdm09 virus: 239, 289 and 391. Mutation at position 222 of HA1 (codon 239) leads to the extension of receptor specificity.Amino acid at position 47 of HA2 (codon 391) is located in the epitope CR6261, which is recognized by the CR6261 antibody. The neutralizing effect of this antibody is associated with HA fusion [16]. According to the molecular modeling data, the mutation E391K was shown to strengthen the interaction between the HA monomers and weaken intrasubunit interactions [17].
Influenza viruses A(H3N2)
Influenza A(H3N2) viruses circulated on the territory of Russian Federation during the entire observed period except the 2009-2010 season when new H1N1 pandemic happened. The frequency of isolation of this (A(H3N2)) virus varied from 10% in 2010-2011 to about 60% in 2008-2009 and 2011-2012 epidemic seasons (Table 1).
Influenza A(H3N2) viruses of 2006-2007 epidemic season were isolated in 13 Regional Basic Laboratories. A detailed study of the antigenic structure of viruses isolated during this epidemic showed no significant antigenic drift when compared with reference strains and viruses of previous years. On the other hand, we did not observe any new variants of influenza A(H3N2) virus. It should be noted that a gradual extinction of similarity between modern isolates and viruses of 2004-2005 epidemic seasons was observed. 2006-2007 strains reacted with antiserum against A/California/07/2004 reference strain with HI titers 4-fold to 16-fold reduced compared with the titer of the homologous virus. The analysis of the interactions of 2006-2007 isolates with antiserum to A/Wisconsin/67/2005 reference strain showed that this vaccine component was relevant and the produced antibodies had broad-spectrum activity: they neutralized the strains isolated in previous years with the HI titers – twofold to fourfold of the homologous titer. Furthermore, isolated viruses could not be divided into distinctive groups according to their reactivity with antiserum neither to the reference strains nor to the epidemic strains. Тhe occurrence of viruses demonstrating good neutralizing activity with antiserum against A/Wisconsin/67/2005 reference strain is also noteworthy.
Isolated influenza A(H3N2) viruses of 2006-2007 epidemic season differed significantly by their genetic characteristics (Fig. 3). However, in this period, we observed A/Wisconsin/67/2005-like viruses with a number of substitutions in HA (T128A, R142G, K173E). Substitution T128A resulted in the loss of potential glycosylation site. Substitutions K173E and R142G, which were found in the strains, isolated in previous years (e.g. A/Johannesburg/33/1999), resulted in the change of the glycoprotein charge. A number of viruses in this group had substitution L157S in the antigenic site B1, substitution N145K in A site and substitution D188E in B2 site. The substitutions S193F (antigenic site B2), N216S (antigenic site D) and V112I, K173E were also noted in circulating viruses. On the other hand, the second group consisted of A/Brisbane/10/2007-like strains, recommended by the WHO for vaccine production in 2008, which have new substitutions G50E and K140I.

The tree was constructed using maximum likelihood method, TN93+G model, bootstrap 1000 replications. Viruses sequenced in the Research Institute of Influenza are marked in bold. ♦ and ◊ – vaccine viruses.
Influenza A(H3N2) viruses were registered in 15 cities of Russia during the 2007-2008 epidemic season, but their share was small in comparison to A(H1N1) and B viruses, accounting for only 15% of all isolates. In the 2008-2009 epidemic season, influenza A(H3N2) viruses constituted the majority of the isolated strains (58.7%) and were observed in 19 cities of different regions of Russia. All of the studied strains were related to A/ Wisconsin/67/2005 reference strain and epidemic isolates of the previous season. Sera to A/Wisconsin/67/2005 and A/Saint Peterburg/71/2007 strains neutralized Russian isolates of 2009 with 2-8-fold reduction compared to the homologous titer.
Summarizing the results of the antigenic analysis of 2006-2009 influenza A(H3N2) viruses, we can conclude that these isolates were mostly homogeneous with small proportions of drift variants. This can be attributed to the lack of or insignificant changes in HA and NA surface proteins of the influenza viruses.
Influenza A(H3N2) viruses of 2007-2009 epidemic seasons were genetically related to the A/ Brisbane/10/2007 vaccine strain (Fig. 3). These strains were characterized by HA amino acid substitutions G50E (antigenic site C) and K140I (antigenic site A) as well as NA substitutions N93D, H150R and Y194I while M1 and M2 proteins did not carry any substitutions. Most of the analyzed 2007 strains carried the rimantadine resistance mutation (S31N) in the M2 protein. Within this genetic clade we found a small separate cluster of viruses, containing specific HA and NA mutations, consisting of a number of Moscow isolates (e.g. A/Moscow/118/2008). The HA sequences were characterized by the substitution K92R, while NA sequences were found to contain two synonymous mutations: 351 A>G (A to G) (in T117 codon) and 408 G>A (G to A) (in Q136 codon).
Almost all M1 sequences of 2008 strains contained an amino acid residue substitution K174R. We did not find any strains related to the new phylogenetic lineage of A/ Perth/16/2009-like viruses among the analyzed strains. Thus, influenza A(H3N2) viruses isolated during 2008-2009 were proved to be the evolutionary continuation of strains of the 2007-2008 epidemic season.
The substitution D147N has been found in the glycosylation site of NA sequences of the majority of A(H3N2) 2008 influenza viruses and all 2009 viruses. It is known that the glycosylation at position 130 of N1 (which corresponds to position 146 in N2) of A/WSN/33 (H1N1) impairs the neuraminidase interaction with plasminogen and reduces the virus pathogenicity [18].
The vast majority of influenza A(H3N2) strains isolated in 2008 carried K174R mutation in M1 protein C-terminal domain, which facilitates the formation of its α-helix conformation [19]. The M2 protein of all studied 2008 strains contains a mutation (S31N) leading to rimantadine resistance.
Influenza A(H3N2) viruses did not circulate during the 2009-2010 epidemic season in Russia and had no epidemiological significance during 2010-2011: sporadic detections of influenza viruses of this subtype were reported in the European part of Russia, and showed a weak activity in Siberia and the Far East. We showed that all viruses isolated in the east of Russia (2006-2013) belonged to A/Victoria/210/2009 genetic group (R261Q) of A/Perth/16/2009 clade (E62K, N144K). The HA of these viruses contains a number of changes in the antigenic site E2. Viruses isolated in the European part of Russia belonged to the A/Perth/10/2010 genetic group of A/Victoria/208/2009 clade and had substitutions in the antigenic site C. All of the studied A(H3N2) strains isolated in 2010-2011 contain a mutation (S31N) in the M gene determining resistance to rimantadine.
All of the studied influenza A(H3N2) viruses of the 2011-2012 epidemic season are related to the A/Stockholm/18/2011 genetic group (V223I) of A/ Victoria/208/2009 clade and contain HA N312S amino acid substitution. These strains could be divided into two subgroups. The first subgroup includes viruses with substitutions N145S in the antigenic site A and A198S in antigenic site B2 (e.g. A/Saint Petersburg/60/2012, A/ Astrakhan/65/2011); second subgroup includes viruses with substitutions S145N, D487N, Q33R, S45N related to the emergence of the potential glycosylation site. Viruses with substitutions T48I, N278K in antigenic site C and T128N, A198P in antigenic site B2 form subgroups 3B and 3C respectively (e.g. A/Kaliningrad/15/2012, A/Saint Petersburg/50/2012).
Analysis of NA sequences of Russian A(H3N2) strains of 2006-2012 did not reveal mutation E119V that leads to oseltamivir resistance.
According to the results of the detailed antigenic analysis, the majority of influenza A(H3N2) strains isolated in 2011-2012 were related to the A/Perth/16/2009 reference strain. These viruses interacted with the appropriate antiserum fourfold reduced compared to the homologous titer. However, we found some strains with the HI titer eightfold reduced in comparison with the homologous titer. Further analysis showed that all of the studied Russian isolates were antigenically close to the viruses of A/Victoria/208/2009 subgroup. These isolates reacted with antibodies against A/Victoria/208/2009 strain up to fourfold decrease of the homologous titer. Nevertheless, we found in this subgroup some strains, which reacted only with eightfold reduction of the homologous titer (e.g. A/ Astrakhan/7/2012). All isolates reacted with antiserum to Russian representative strain A/SaintPetersburg/10/2012 1-4 fold less compared to the homologous titer. The latter strains differed from the reference strains in their interaction with antisera to viruses isolated in 2009 (A/ Perth/16/2009,A/Victoria/208/2009,A/Victoria/210/2009). Although, like other strains of the 2012-2013 season, they reacted poorly with antisera to the new reference viruses A/Victoria/361/2011 selected by the WHO experts to be included in the vaccine for the 2012-2013 season [20]. This proves that influenza A(H3N2) viruses had a unique path of evolution in Russia.
The phylogenetic analysis of the HA gene of influenza A(H3N2) viruses isolated in the 2011-2012 epidemic period showed that the vast majority of strains belonged to A/Victoria/208/2009 clade. They were related to the A/Victoria/361/2011-like group and could be divided into two subgroups: the first subgroup includes viruses with substitutions N145S (antigenic site A) and D487N (e.g. strains A/Saint Petersburg/01/2012, A/ Petrozavodsk/01/2012); the second subgroup includes viruses with substitutions Q33R, S45N (emergence of a new potential glycosylation site), T48I, N278K (antigenic site C) (e.g. A/Omsk/01/2012 and A/Novosibirsk/08/2012). It should be noted that the strains A/Omsk/01/2012, A/ Novosibirsk/08/2012, and A/Novosibirsk/09/2012 have the amino acid substitution A198P in the antigenic site B2. The other 3 strains, isolated in the same epidemic period, were related to the A/Stockholm/18/2011-like group (Fig. 3): they have amino acid substitutions V223I and N312S unlike A/Victoria/208/2009
None of the strains analyzed in 2012 belonged to the A/Perth/10/2010-group A/Victoria/208/2009-clade with characteristic substitutions D53N, Y94H, I230V, and E280A.
All of the isolates of the season 2012-2013 interacted poorly in HI test with the antiserum raised against egg-derived A/Victoria/361/2011 strain, as well as with antiserum to the egg-derived reference strains of 2012 such as A/South Australia/30/2012 and A/Ohio/2/2012. At the same time, the majority of 2011-2013 isolates revealed high HI titers with antiserum raised against cell-grown reference virus A/Victoria/361/2011.
The HA phylogenetic analysis of influenza A(H3N2) strains isolated in the 2012-2013 epidemic period showed that all of the strains were A/Victoria/361/2011-like (subgroup 3C of A/Victoria/208/2009 clade), since their structure was defined by amino acid changes S45N, T48I, S145N, A198S, V223I and N312S. The majority of these strains contained substitution T128A in HA1 presumably leading to the loss of glycosylation site at position 126. Substitutions in antigenic site A (N121D, S124N, I140M) are common for these strains.
All influenza A strains isolated during the last years of the reported period were found to be susceptible to NA inhibitors and resistant to adamantane antivirals. None of them carried H275Y substitution in NA, and all had S31N substitution in M2.
Influenza B viruses
Influenza B viruses of both Yamagata and Victoria lineages circulated in Russia in the period from 2006 to 2013. The vast majority of influenza B viruses belonged to the Victoria lineage. The occurrence of B/ Malaysia/2506/04-like strains was observed in Russia during 2006-2007 epidemic season. All isolates reacted with antiserum raised against this strain onefold to fourfold of the homologous titer. However, the emergence of viruses belonging to B/Brisbane/60/2008-like strains of Victoria lineage was registered in 2008-2009 season. Circulation of these viruses continues to the present day. Influenza activity of the Yamagata lineage was not that intense in the observed period. Viruses of Yamagata lineage clearly prevailed over the Victoria one only in 2007-2008 season: 98% of all isolated influenza B viruses were B/Florida/4/2006-like. The next occurrence of Yamagata group viruses was registered only in 2011-2012 season when viruses, related to the new vaccine strain B/ Wisconsin/1/2010 made up 30% of all isolates.
We performed the phylogenetic analysis of HA gene fragments of influenza B strains isolated in different regions of the Russian Federation in the 2011-2012 epidemic season (Fig. 4). The analysis of influenza B strains isolated in previous seasons showed that all of them belonged to the Victoria lineage clade III-ii or B/ Brisbane/60/2008-like strains. Considering that the influenza B viruses of the Victoria lineage circulated in Russia during 3 epidemic seasons, we could expect the emergence of influenza B of the Yamagata line in Russia in the epidemic of 2011-2012.

The tree was constructed using maximum likelihood method, HKY+G model, bootstrap 1000 replications. Viruses sequenced in the Research Institute of Influenza are shown in bold. ♦ – vaccine viruses.
As expected, influenza B of both the Victoria and Yamagata lineages were found during 2012-2013 epidemic season in Russia. The HA phylogenetic analysis of 4 strains, belonging to Yamagata lineage, isolated in 2012 showed that they all are related to the B/Bangladesh/3333/2007-like group with substitutions N165Y and S150I in the antigenic sites BB2 and BA, respectively (Fig. 5). Furthermore, the strains isolated in Russia in 2012 (for example, B/ Novosibirsk/01/2012) have HA substitutions K298E, E312R, and N116K (antigenic site BC). The 11 analyzed strains of 2012 belonged to the B/Brisbane/60/2008- like strains of the Victoria lineage and have a number of amino acid substitutions: N75K (antigenic site BE), N165K (antigenic site BB2) and S172P (Fig. 4). In addition, all the Russian isolates had the following substitutions in the HA: L58P (antigenic site BE) and V146I (antigenic site BA) in the Victoria lineage strains and T181K (antigenic site BD) in the Yamagata lineage strains.
In 2012-2013 season, the simultaneous circulation of influenza B strains of Yamagata and Victoria lineages was also observed with the prevalence of Yamagata lineage (87%) (Table 1).
2012-2013 Yamagata lineage strains were similar to the reference strain B/Wisconsin/01/2010, which was included in the vaccine composition for this season (2012-2013). These viruses were also similar to the Russian isolates of 2012-2013 and to the new reference-strain B/Massachusetts/2/2012, proposed by the WHO to be included into the seasonal vaccine for the Northern Hemisphere for the season 2013-2014. At the same time, they have some relationship to the earlier strain B/Bangladesh/3333/2007. Antigenic cartography of influenza viruses B of Yamagata lineage (data not shown) has demonstrated a gradual drift of Russian isolates from the reference strains B/Florida/07/2004 and B/Florida/04/2006 (strains isolated during 2005-2008) to the strains similar to the reference virus B/ Wisconsin/01/2010 and its Russian analogue B/Saint Petersburg/24/2012. At the same time, the reference strain B/Bangladesh/3333/2007 has an intermediate position on two-dimensional cartogram between the two above mentioned groups.
According to the results of antigenic analysis, all of the B/Victoria lineage strains were similar to the vaccine reference strain B/Brisbane/60/08 and reacted with the antiserum to that strain up to 4- fold of the homological titer. All isolates of the 2012-2013 season also reacted well in the HI test with antisera to the Russian strains isolated in 2009-2013. Some of them were chosen as “local” reference viruses of the Research Institute of Influenza.
All strains of B/Victoria lineage isolated in 2012-2013 season fell into clade 1A and have common substitution K209T in HA1 subunit unlike B/Victoria strains isolated in 2012 that fell into clades 1A and 1B which have N197K and T199A substitutions in HA receptor-binding site (Fig. 4). No intra-clade reassortant HA-1A/NA-4 was identified in this season. B/Yamagata lineage strains of clade 2 (B/Brisbane/03/2007-like) contained amino acid changes R48K (antigenic site BC), I150S (antigenic site BA), Y165N (antigenic site BB2), T181A (antigenic site BD). Only two of these strains belonged to clade 3 (B/ Wisconsin/01/2010-like) (Fig. 5).

The tree was constructed using maximum likelihood method, HKY+G model, bootstrap 1000 replications. Viruses sequenced in the Research Institute of Influenza are shown in bold. ♦ and ◊ – vaccine viruses.
No changes leading to resistance to oseltamivir or zanamivir were found in influenza B strains isolated until 2013.
Thus, we studied the antigenic and genetic diversity of influenza A and B viruses that circulated in Russia in 2006-2013. The analysis of this data proves that in this period significant changes occurred in the genetic structure of influenza viruses isolated on the territory of Russia, as well as in their sensitivity to antiviral drugs and phylogenetic affiliation. In addition, since the summer of 2009, a new pandemic influenza virus A(H1N1)pdm09 began to spread in Russia.
The identification of codons under the pressure of positive selection in silico in the genes encoding surface proteins of influenza viruses might be very informative for the prediction of their antigenic drift and evolutionary variability.
Continuous monitoring of the mutational variability and phylogenetic analysis of circulating strains play an important role in the selection of vaccine strains for influenza specific prevention and antiviral drugs. Phylogenetic analysis is also a powerful tool for identifying new reassortants that could have epidemic or pandemic potential.