- Record: found
- Abstract: found
- Article: found
Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle
Read this article at
Abstract
Background
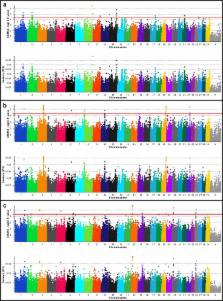
Single nucleotide polymorphism (SNP) arrays for domestic cattle have catalyzed the identification of genetic markers associated with complex traits for inclusion in modern breeding and selection programs. Using actual and imputed Illumina 778K genotypes for 3887 U.S. beef cattle from 3 populations (Angus, Hereford, SimAngus), we performed genome-wide association analyses for feed efficiency and growth traits including average daily gain (ADG), dry matter intake (DMI), mid-test metabolic weight (MMWT), and residual feed intake (RFI), with marker-based heritability estimates produced for all traits and populations.
Results
Moderate and/or large-effect QTL were detected for all traits in all populations, as jointly defined by the estimated proportion of variance explained (PVE) by marker effects (PVE ≥ 1.0%) and a nominal P-value threshold ( P ≤ 5e-05). Lead SNPs with PVE ≥ 2.0% were considered putative evidence of large-effect QTL ( n = 52), whereas those with PVE ≥ 1.0% but < 2.0% were considered putative evidence for moderate-effect QTL ( n = 35). Identical or proximal lead SNPs associated with ADG, DMI, MMWT, and RFI collectively supported the potential for either pleiotropic QTL, or independent but proximal causal mutations for multiple traits within and between the analyzed populations. Marker-based heritability estimates for all investigated traits ranged from 0.18 to 0.60 using 778K genotypes, or from 0.17 to 0.57 using 50K genotypes (reduced from Illumina 778K HD to Illumina Bovine SNP50). An investigation to determine if QTL detected by 778K analysis could also be detected using 50K genotypes produced variable results, suggesting that 50K analyses were generally insufficient for QTL detection in these populations, and that relevant breeding or selection programs should be based on higher density analyses (imputed or directly ascertained).
Conclusions
Fourteen moderate to large-effect QTL regions which ranged from being physically proximal (lead SNPs ≤ 3Mb) to fully overlapping for RFI, DMI, ADG, and MMWT were detected within and between populations, and included evidence for pleiotropy, proximal but independent causal mutations, and multi-breed QTL. Bovine positional candidate genes for these traits were functionally conserved across vertebrate species.
Related collections
Most cited references144
- Record: found
- Abstract: found
- Article: not found
An efficient multi-locus mixed model approach for genome-wide association studies in structured populations
- Record: found
- Abstract: found
- Article: not found
Development and Characterization of a High Density SNP Genotyping Assay for Cattle
- Record: found
- Abstract: found
- Article: not found