- Record: found
- Abstract: found
- Article: found
Cognitive deficits and Alzheimer-like neuropathological impairments during adolescence in a rat model of type 2 diabetes mellitus
Read this article at
Abstract

Abstract
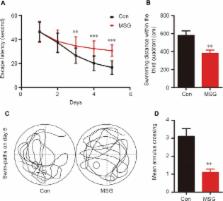
Numerous studies have shown that many patients who suffer from type 2 diabetes mellitus exhibit cognitive dysfunction and neuronal synaptic impairments. Therefore, growing evidence suggests that type 2 diabetes mellitus has a close relationship with occurrence and progression of neurodegeneration and neural impairment in Alzheimer's disease. However, the relationship between metabolic disorders caused by type 2 diabetes mellitus and neurodegeneration and neural impairments in Alzheimer's disease is still not fully determined. Thus, in this study, we replicated a type 2 diabetic animal model by subcutaneous injection of newborn Sprague-Dawley rats with monosodium glutamate during the neonatal period. At 3 months old, the Barnes maze assay was performed to evaluate spatial memory function. Microelectrodes were used to measure electrophysiological function in the hippocampal CA1 region. Western blot assay was used to determine expression levels of glutamate ionotropic receptor NMDA type subunit 2A (GluN2A) and GluN2B in the hippocampus. Enzyme-linked immunosorbent assay was used to determine levels of interleukin-1β, tumor necrosis factor α, and interleukin-6 in the hippocampus and cerebral cortex, as well as hippocampal amyloid beta (Aβ) 1–40 and Aβ 1–42 levels. Our results showed that in the rat model of type 2 diabetes mellitus caused by monosodium glutamate exposure during the neonatal period, latency was prolonged and the number of errors increased in the Barnes maze. Further, latency was increased and time in the escape platform quadrant shortened. Number of times crossing the platform was also reduced in the Morris water maze. After high frequency stimulation of the hippocampus, synaptic transmission was inhibited, expression of GluN2A and GluN2B were decreased in the hippocampus, expression of interleukin 1β, interleukin 6, and tumor necrosis factor α was increased in the hippocampus and cortex, and levels of Aβ 1–40 and Aβ 1–42 were increased in the hippocampus. These findings confirm that type 2 diabetes mellitus induced by neonatal monosodium glutamate exposure results in Alzheimer-like neuropathological changes and further causes cognitive deficits and neurodegeneration in young adulthood.
Related collections
Most cited references55
- Record: found
- Abstract: found
- Article: not found