- Record: found
- Abstract: found
- Article: found
Reference-Free Population Genomics from Next-Generation Transcriptome Data and the Vertebrate–Invertebrate Gap
Read this article at
Abstract
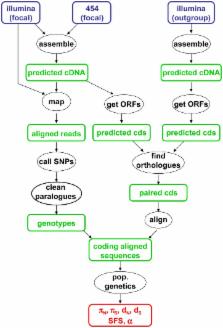
In animals, the population genomic literature is dominated by two taxa, namely mammals and drosophilids, in which fully sequenced, well-annotated genomes have been available for years. Data from other metazoan phyla are scarce, probably because the vast majority of living species still lack a closely related reference genome. Here we achieve de novo, reference-free population genomic analysis from wild samples in five non-model animal species, based on next-generation sequencing transcriptome data. We introduce a pipe-line for cDNA assembly, read mapping, SNP/genotype calling, and data cleaning, with specific focus on the issue of hidden paralogy detection. In two species for which a reference genome is available, similar results were obtained whether the reference was used or not, demonstrating the robustness of our de novo inferences. The population genomic profile of a hare, a turtle, an oyster, a tunicate, and a termite were found to be intermediate between those of human and Drosophila, indicating that the discordant genomic diversity patterns that have been reported between these two species do not reflect a generalized vertebrate versus invertebrate gap. The genomic average diversity was generally higher in invertebrates than in vertebrates (with the notable exception of termite), in agreement with the notion that population size tends to be larger in the former than in the latter. The non-synonymous to synonymous ratio, however, did not differ significantly between vertebrates and invertebrates, even though it was negatively correlated with genetic diversity within each of the two groups. This study opens promising perspective regarding genome-wide population analyses of non-model organisms and the influence of population size on non-synonymous versus synonymous diversity.
Author Summary
The analysis of genomic variation between individuals of a given species has so far been restricted to a small number of model organisms, such as human and fruitfly, for which a fully sequenced, well-annotated reference genome was available. Here we show that, thanks to next-generation high-throughput sequencing technologies and appropriate genotype-calling methods, de novo population genomic analysis is possible in absence of a reference genome. We characterize the genomic level of neutral and selected polymorphism in five non-model animal species, two vertebrates and three invertebrates, paying particular attention to the treatment of multi-copy genes. The analyses demonstrate the influence of population size on genetic diversity in animals, the two vertebrates (hare, turtle) and the social insect (termite) being less polymorphic than the two marine invertebrates (oyster, tunicate) in our sample. Interestingly, genomic indicators of the efficiency of natural selection, both purifying and adaptive, did not vary in a simple, predictable way across organisms. These results prove the value of a diversified sampling of species when it comes to understand the determinants of genome evolutionary dynamics.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
Population genomics of domestic and wild yeasts
- Record: found
- Abstract: found
- Article: found
A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data
- Record: found
- Abstract: found
- Article: not found