- Record: found
- Abstract: found
- Article: found
PKD1-associated autosomal dominant polycystic kidney disease with glomerular cysts presenting with nephrotic syndrome caused by focal segmental glomerulosclerosis
Read this article at
Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) may manifest non-nephrotic range proteinuria, but is rarely complicated with nephrotic syndrome. Limited number of reports describe the histology of ADPKD with nephrotic syndrome in detail.
Case presentation
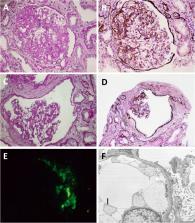
We encountered a 23-year-old man with polycystic kidney disease (PKD) with small kidney volume and nephrotic syndrome, which eventually progressed to end-stage renal disease. Renal histology showed typical focal segmental glomerulosclerosis and remarkable glomerular cyst formation, but did not reveal tubular cysts. PKD1 mutation was detected in him and his father, who also had PKD with small kidney volume.
Conclusions
In contrast to tubular cysts which develop along ADPKD progression, glomerular cysts may likely be associated with ADPKD with slower volume progression manifesting small kidney volume. Although previous investigations report that ADPKD with smaller kidney volume is attributed to slower decline in renal function, coexistence of nephrotic-range proteinuria implies complication of other glomerular diseases and needs histological evaluation since it may lead to poor renal outcome.
Related collections
Most cited references8
- Record: found
- Abstract: found
- Article: not found
Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials.
- Record: found
- Abstract: found
- Article: not found
From segmental glomerulosclerosis to total nephron degeneration and interstitial fibrosis: a histopathological study in rat models and human glomerulopathies.
- Record: found
- Abstract: found
- Article: not found