Introduction The human immune-mediated diseases are the result of aberrant immune responses. These immune responses may lead to chronic inflammation and tissue destruction, often targeting a specific organ site. The outcome of this process is immune-mediated inflammatory and autoimmune disease, affecting approximately 5% of the population [1]. Extensive clinical and epidemiologic observations have shown that immune-mediated inflammatory and autoimmune diseases can occur either in the same individual or in closely related family members. This clustering of multiple diseases appears more frequently than expected if disease processes were independent. As each of the immune-mediated inflammatory and autoimmune diseases has strong genetic influences on disease risk [2]–[7], the observed clustering of multiple diseases could be due to an overlap in the causal genes and pathways [8], [9]. The patterns of clustering of diseases across the population are complex [10] – each disease has a prevalence between 0.01%–3%, so direct assessment of co-aggregation within individuals or families does not result in the very large samples required for genetic or epidemiological investigation. Thus it is unsurprising that to date, these observations have yet to be translated into determinants of the shared molecular etiologies of disease. Recent GWA studies in immune-mediated and autoimmune diseases have identified 140 regions of the genome with statistically significant and robust evidence of presence of disease susceptibility loci. A subset of these loci have been shown to modulate risk of multiple diseases [3], [6], [11]–[14]. In addition, there is evidence that loci predisposing to one disease can have effects on risk of a second disease [15], although the risk allele for one disease may not be the same as for the second [16]. Together, these observations support the hypothesis of a common genetic basis of immune-mediated and autoimmune diseases [17]. There is now the ability to estimate both the number of loci contributing to risk of multiple diseases and the spectrum of diseases that each locus influences. In addition, grouping variants by the diseases they influence should provide insight into the specific biological processes underlying co-morbidity and disease risk. In this report, we systematically investigate the genetic commonality in immune-mediated inflammatory and autoimmune diseases by examining the contributions of associated genomic risk regions in seven diseases: celiac disease (CeD), Crohn's disease (CD), multiple sclerosis (MS), psoriasis (Ps), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and type 1 diabetes (T1D). We find that nearly half of loci identified in GWAS studies of an individual disease influence risk to at least two diseases, arguing for a genetic basis to co-morbidity. We also find several variants with opposing risk profiles in different diseases. Supporting the idea that common patterns of association implicate shared biological processes, we further demonstrate that loci clustered by the pattern of diseases they affect harbor genes encoding interacting proteins at a much higher rate than by chance. These results suggest that multi-phenotype mapping will identify the molecular mechanisms underlying co-morbid immune-mediated inflammatory and autoimmune diseases. Results We first test our hypothesis of common genetic determinants by examining evidence of association of genetic variants in known immune-mediated and autoimmune disease susceptibility loci to multiple disease phenotypes. We collated a list of 140 single nucleotide polymorphisms (SNPs) representing reported associations to at least one immune-mediated disease at genome-wide significance levels. Where data for the reported SNP itself were not available in our GWA studies (Table 1), we chose a proxy in high linkage disequilibrium to the reported marker (r2 >0.9 in HapMap/CEU). We did not consider SNPs in the human Major Histocompatibility Complex (MHC) from this analysis, as its role in many of these diseases is well-established and the classically associated alleles in the HLA region are not well captured by SNPs [18]. We were able to acquire data for either the reported SNP or a good proxy in 107 of 140 cases, and assembled genotype test summaries for these from previously described GWA studies representing over 26,000 disease cases (Table 1). 10.1371/journal.pgen.1002254.t001 Table 1 Participating studies. Disease Cases Controls Reference Celiac disease 3796 8154 22 Crohn's disease 3230 4829 1 Multiple sclerosis 2624 7220 4 Psoriasis 1359 1400 5 Rheumatoid arthritis 5539 20169 6 Systemic Lupus Erythematosus 1963 4329 23 Type 1 diabetes 7514 9045 24 Data were collated for seven phenotypes from meta-analyses incorporating all known genome-wide association studies. SLE is the exception as no comprehensive meta-analysis has yet been published; data were instead obtained from a recent meta-analysis including some, but not all, known genome-wide association studies. Note that controls overlap in some cases due to the use of common shared sample genotypes. We have developed a cross-phenotype meta-analysis (CPMA) statistic to assess association across multiple phenotypes. The CPMA statistic determines evidence for the hypothesis that each independent SNP has multiple phenotypic associations. Support for this hypothesis would be shown by deviations from expected uniformity of the distribution of association p-values, indicative of multiple associations. The likelihood of the observed rate of exponential decay of −log10(p) is calculated and compared to the null expectation (the decay rate should be unity) as a likelihood ratio test (see Materials and Methods for details). This CPMA statistic has one degree of freedom, as it measures a deviation in p-value behavior instead of testing all possible combinations of diseases for association to each SNP. A total of 47 of the 107 SNPs tested have evidence of association to multiple diseases (SNP-wise PCPMA 0.9) to represent the region. Cross-phenotype meta-analysis Our CPMA analysis relies on the expected distribution of p-values for each SNP across diseases. Under the null hypothesis of no additional associations beyond those already known, we expect association values to be uniformly distributed and hence -ln(p) to be exponentially decaying with a decay rate λ = 1. We calculate the likelihood of the observed and expected values of λ and express these as a likelihood ratio test: This statistic therefore measures the likelihood of the null hypothesis given the data; we can reject the null hypothesis if sufficient evidence to the contrary is present. We note that, because we only estimate a single parameter, our test is asymptotically distributed as . This gives us more statistical power than relying on strategies combining association statistics, which would consume multiple degrees of freedom. SNP–SNP distance calculation and clustering To compare the patterns of association for multi-phenotype SNPs we first calculate SNP-SNP distances and then use hierarchical clustering on that distance matrix to assess relative relationships between SNP association patterns. Calculating distances based directly on p values or the underlying association statistics is problematic, as each contributing study has slightly different sample sizes and therefore different statistical power to detect associations. Thus, distance functions based on numeric data – which incorporate magnitude differences between observations – would be biased if studies have systematically different data. Normalization procedures can account for such systematic differences but may fail to remove all bias. To reduce the impact such systematic irregularities might have on our comparison, we bin associations into informal “levels of evidence” categories. We define four classes (1

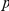 -values
-values
 ,
,
 , and
, and
 , respectively). We also report associations with rs4915077 near
VAV3 (
, respectively). We also report associations with rs4915077 near
VAV3 (
 -value
-value
 ) and rs925489 near
FOXE1 (
) and rs925489 near
FOXE1 (
 -value
-value
 ).
VAV3 is involved in immune function, and
FOXE1 and
PTPN22 have previously been associated with hypothyroidism. Although the HLA class I region
and
SH2B3 have previously been linked with a number of autoimmune diseases, this is the first
report of their association with thyroid disease. The
VAV3 association is also novel. We also show suggestive evidence of association for hypothyroidism
with a SNP in the HLA class II region (independent of the other HLA association) as
well as SNPs in
CAPZB,
PDE8B, and
CTLA4.
CAPZB and
PDE8B have been linked to TSH levels and
CTLA4 to a variety of autoimmune diseases. These results suggest heterogeneity in the genetic
etiology of hypothyroidism, implicating genes involved in both autoimmune disorders
and thyroid function. Using a genetic risk profile score based on the top association
from each of the five genome-wide significant regions in our study, the relative risk
between the highest and lowest deciles of genetic risk is 2.0.
).
VAV3 is involved in immune function, and
FOXE1 and
PTPN22 have previously been associated with hypothyroidism. Although the HLA class I region
and
SH2B3 have previously been linked with a number of autoimmune diseases, this is the first
report of their association with thyroid disease. The
VAV3 association is also novel. We also show suggestive evidence of association for hypothyroidism
with a SNP in the HLA class II region (independent of the other HLA association) as
well as SNPs in
CAPZB,
PDE8B, and
CTLA4.
CAPZB and
PDE8B have been linked to TSH levels and
CTLA4 to a variety of autoimmune diseases. These results suggest heterogeneity in the genetic
etiology of hypothyroidism, implicating genes involved in both autoimmune disorders
and thyroid function. Using a genetic risk profile score based on the top association
from each of the five genome-wide significant regions in our study, the relative risk
between the highest and lowest deciles of genetic risk is 2.0.
