- Record: found
- Abstract: found
- Article: found
HOMINID: a framework for identifying associations between host genetic variation and microbiome composition
Read this article at
Abstract
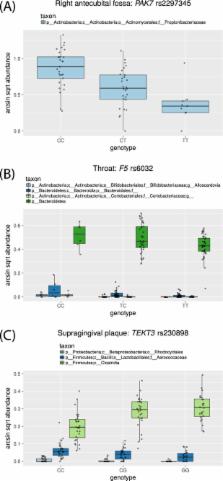
Recent studies have uncovered a strong effect of host genetic variation on the composition of host-associated microbiota. Here, we present HOMINID, a computational approach based on Lasso linear regression, that given host genetic variation and microbiome taxonomic composition data, identifies host single nucleotide polymorphisms (SNPs) that are correlated with microbial taxa abundances. Using simulated data, we show that HOMINID has accuracy in identifying associated SNPs and performs better compared with existing methods. We also show that HOMINID can accurately identify the microbial taxa that are correlated with associated SNPs. Lastly, by using HOMINID on real data of human genetic variation and microbiome composition, we identified 13 human SNPs in which genetic variation is correlated with microbiome taxonomic composition across body sites. In conclusion, HOMINID is a powerful method to detect host genetic variants linked to microbiome composition and can facilitate discovery of mechanisms controlling host-microbiome interactions.
Related collections
Most cited references8
- Record: found
- Abstract: not found
- Article: not found
Stability selection
- Record: found
- Abstract: found
- Article: not found
Association mapping in structured populations.
- Record: found
- Abstract: found
- Article: not found