- Record: found
- Abstract: found
- Article: found
Pivotal role of the muscle-contraction pathway in cryptorchidism and evidence for genomic connections with cardiomyopathy pathways in RASopathies

Read this article at
Abstract
Background
Cryptorchidism is the most frequent congenital disorder in male children; however the genetic causes of cryptorchidism remain poorly investigated. Comparative integratomics combined with systems biology approach was employed to elucidate genetic factors and molecular pathways underlying testis descent.
Methods
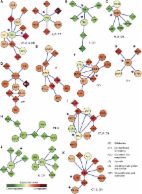
Literature mining was performed to collect genomic loci associated with cryptorchidism in seven mammalian species. Information regarding the collected candidate genes was stored in MySQL relational database. Genomic view of the loci was presented using Flash GViewer web tool (http://gmod.org/wiki/Flashgviewer/). DAVID Bioinformatics Resources 6.7 was used for pathway enrichment analysis. Cytoscape plug-in PiNGO 1.11 was employed for protein-network-based prediction of novel candidate genes. Relevant protein-protein interactions were confirmed and visualized using the STRING database (version 9.0).
Results
The developed cryptorchidism gene atlas includes 217 candidate loci (genes, regions involved in chromosomal mutations, and copy number variations) identified at the genomic, transcriptomic, and proteomic level. Human orthologs of the collected candidate loci were presented using a genomic map viewer. The cryptorchidism gene atlas is freely available online: http://www.integratomics-time.com/cryptorchidism/. Pathway analysis suggested the presence of twelve enriched pathways associated with the list of 179 literature-derived candidate genes. Additionally, a list of 43 network-predicted novel candidate genes was significantly associated with four enriched pathways. Joint pathway analysis of the collected and predicted candidate genes revealed the pivotal importance of the muscle-contraction pathway in cryptorchidism and evidence for genomic associations with cardiomyopathy pathways in RASopathies.
Conclusions
The developed gene atlas represents an important resource for the scientific community researching genetics of cryptorchidism. The collected data will further facilitate development of novel genetic markers and could be of interest for functional studies in animals and human. The proposed network-based systems biology approach elucidates molecular mechanisms underlying co-presence of cryptorchidism and cardiomyopathy in RASopathies. Such approach could also aid in molecular explanation of co-presence of diverse and apparently unrelated clinical manifestations in other syndromes.
Related collections
Most cited references77
- Record: found
- Abstract: found
- Article: found
Network-based classification of breast cancer metastasis
- Record: found
- Abstract: found
- Article: not found
An atlas of combinatorial transcriptional regulation in mouse and man.

- Record: found
- Abstract: found
- Article: found