- Record: found
- Abstract: found
- Article: found
Escape of Actively Secreting Shigella flexneri from ATG8/LC3-Positive Vacuoles Formed during Cell-To-Cell Spread Is Facilitated by IcsB and VirA
Read this article at
ABSTRACT
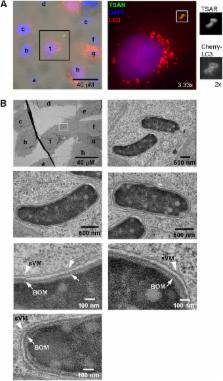
The enteropathogenic bacterium Shigella flexneri uses a type 3 secretion apparatus (T3SA) to transfer proteins dubbed translocators and effectors inside host cells, inducing bacterial uptake and subsequent lysis of the entry vacuole. Once in the cytoplasm, the outer membrane protein IcsA induces actin polymerization, enabling cytoplasmic movement and cell-to-cell spread of bacteria. During this infectious process, S. flexneri is targeted by ATG8/LC3. The effector IcsB was proposed to inhibit LC3 recruitment by masking a region of IcsA recognized by the autophagy pathway component ATG5. The effector VirA, a GTPase-activating protein (GAP) for Rab1, was also shown to prevent LC3 recruitment. However, the context of LC3 recruitment around S. flexneri is not fully understood. Here, we show that LC3 is recruited specifically around secreting bacteria that are still present in vacuoles formed during entry and cell-to-cell spread. While LC3 recruitment occurs around a small proportion of intracellular wild-type bacteria, the icsB, virA, and icsB virA mutants display incremental defaults in escape from LC3-positive vacuoles formed during cell-to-cell spread. Our results indicate that IcsB and VirA act synergistically to allow bacteria to escape from LC3-positive vacuoles by acting at or in the immediate vicinity of the vacuole membrane(s). We also demonstrate that LC3 is recruited around bacteria still present in the single-membrane entry vacuole, in a manner akin to that seen with LC3-associated phagocytosis. Our results indicate that LC3 recruitment occurs around bacteria still, or already, in membrane compartments formed during entry and cell-to-cell spread, and not around bacteria free in the cytoplasm.
IMPORTANCE
The targeting of S. flexneri by LC3 is a classic example of the targeting of foreign cytoplasmic particles by autophagy (so-called “xenoautophagy”). It is often assumed that LC3 is recruited around bacteria present in the cytoplasm through the formation of canonical double-membrane autophagosomes. Our results indicate that LC3 is recruited around the entry vacuole composed of a single membrane as in the case of LC3-associated phagocytosis. Effectors IcsB and VirA had been implicated in the blocking of LC3 recruitment, but it was not known if they acted on the same or distinct LC3-recruiting pathways. Our results indicate that LC3 is recruited exclusively around bacteria present in vacuoles formed during entry and cell-to-cell spread and that both IcsB and VirA intervene at the latter stage to facilitate bacterial escape. Our report reconciles several findings and may have broad implications for our understanding of the specific targeting of bacterial pathogens by LC3.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria.
- Record: found
- Abstract: found
- Article: not found