- Record: found
- Abstract: found
- Article: not found
Targeted DNA Demethylation and Endogenous Gene Activation Using Programmable TALE-TET1 Fusions
research-article
Morgan L Maeder
1
,
2
,
3 ,
James F Angstman
1
,
2
,
4 ,
Marcy E Richardson
5 ,
Samantha J Linder
1
,
2 ,
Vincent M Cascio
1
,
2 ,
Shengdar Q Tsai
1
,
2
,
6 ,
Quan H Ho
1
,
2 ,
Jeffry D Sander
1
,
2
,
6 ,
Deepak Reyon
1
,
2
,
6 ,
Bradley E Bernstein
1
,
6
,
7
,
8 ,
Joseph F Costello
9 ,
Miles F Wilkinson
5
,
10 ,
J Keith Joung
1
,
2
,
3
,
6
,
*
09 October 2013
Read this article at

There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Recent genomewide studies have defined cell type-specific patterns of DNA methylation
1
, a modification known to be important for regulating gene expression in both normal
development
2
and disease
3
states. However, determining the functional significance of specific methylation events
remains a challenging problem due to the lack of targeted methodologies for removing
such modifications. Here we describe an approach for efficient targeted demethylation
of specific CpGs in human cells using fusions of engineered transcription activator-like
effector (TALE) repeat arrays and the TET1 hydroxylase catalytic domain. Using these
TALE-TET1 fusions, we demonstrate that modification of certain critical methylated
promoter CpG positions can be associated with substantial increases in endogenous
human gene expression. Our results delineate a general strategy for understanding
the functional significance of specific CpG methylation marks in the context of endogenous
gene loci and validate new programmable DNA demethylation reagents with broad potential
utility for research and therapeutic applications.
Methylation of DNA at cytosine bases is an important mechanism widely used to regulate
gene expression and transposable elements in higher eukaryotic organisms
4
. Regions of hypermethylated DNA in mammalian cells are often associated with silenced,
inactive chromatin whereas regions of hypomethylated DNA are often associated with
expressed genes and open chromatin
1,5
. In mammalian cells, the generation of methylated cytosine (5mC) is catalyzed and
maintained by DNA methyltransferases (DNMTs) primarily at CpG dinucleotides
6
. One pathway of active 5mC demethylation is initiated by the ten-eleven translocation
(TET) family of proteins, enzymes that catalyze the oxidation of 5mC to 5-hydroxymethylcytosine
(5hmC), a critical step that appears to be important for ultimate removal of the methyl
mark
7–13
.
Defining the causal effects of specific CpG methylation events has remained challenging
due to the lack of targeted methods for converting 5mC to unmethylated cytosine in
living cells. Currently, only non-specific approaches exist for removing methyl groups
from CpGs. For example, the cytidine analog 5-aza-2’-deoxycytidine (decitabine), an
inhibitor of DNMTs, has been widely used to study the effects of demethylation on
specific gene promoters. However, decitabine leads to global demethylation of CpGs
in cells, making it difficult to definitively establish causal effects. Here we sought
to specifically demethylate CpGs in a targeted fashion at endogenous genes by fusing
the hydroxylase activity of the human TET1 protein to engineered TALE repeat arrays
with programmable DNA-binding specificities. Customized TALE repeat arrays make an
attractive platform for directing TET1 activity because monomeric proteins that bind
to nearly any target DNA sequence of interest can be robustly made by simple and rapid
assembly of individual repeat domains with known single base specificities
14
.
In initial experiments, we defined the architecture of a TALE-TET1 fusion protein
that could mediate efficient targeted conversion of 5mC to 5hmC at specific CpGs with
resulting subsequent demethylation in human cells. To do this, we fused TALE repeat
arrays engineered to bind two different sites in the human KLF4 gene with either full-length
human TET1 or its catalytic domain (CD) (Figs. 1a, 1b, 1c; Methods). We then tested
whether these four proteins could demethylate CpGs adjacent to the TALE binding sites
in human K562 cells using a bisulfite sequencing protocol that utilizes high-throughput
next-generation sequencing to generate more than 10,000 sequencing reads per sample
(Methods, Supplementary Results, and Supplementary Fig. 1). For both KLF4 target sites,
we found that TALE fusions bearing the TET1 CD domain induced significantly greater
decreases in methylation of CpGs proximal to the TALE binding site than those bearing
the full-length TET1 protein (Fig. 1d and 1e; Methods). For example, one of the TALE-TET1CD
fusion proteins reduced the methylation of CpGs located 10 and 16 bp from the 3’ boundary
of the TALE binding site by 21% and 30%, respectively, with similar levels of demethylation
observed on both DNA strands (Supplementary Fig. 2). Lengthening the linker between
the TALE repeat array and the TET1 CD did not appreciably alter demethylation efficiencies
observed (Supplementary Fig. 3). Therefore, all subsequent experiments used TALE-TET1CD
proteins with a short GGGS linker (hereafter referred to as simply “TALE-TET1” fusion
proteins). Control fusion proteins bearing a TALE repeat array targeted to an unrelated
EGFP reporter gene sequence did not demethylate CpGs in the KLF4 intron (Figs. 1d
and 1e), demonstrating that demethylation requires specific binding to the target
locus by the TALE repeats and is not due simply to overexpression of proteins harboring
TET1 hydroxylase activity. Based on a dose-response experiment, which showed increased
levels of demethylation in cells transfected with greater amounts of plasmid encoding
a TALE-TET1 protein, we identified optimal transfection conditions that maximized
both CpG demethylation and cell viability (Supplementary Fig. 4).
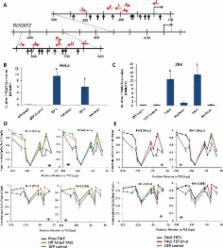
We next determined whether TALE-TET1-induced demethylation of CpGs in human promoters
might induce expression changes in proximal endogenous genes. The RHOXF2 /2B homeobox
gene (hereafter referred to as simply RHOXF2) is expressed primarily in male germ
cells
15
. Plasmid-based reporter gene studies using decitabine have demonstrated that RHOXF2
expression in non-germ cells is strongly repressed by DNA methylation (ref.
16
and M. Richardson et al., manuscript submitted). We engineered eleven TALE-TET1 proteins
(hereafter referred to as RH-1 through RH-11) targeted to sites that lie in close
proximity to a total of 18 different CpGs in the RHOXF2 promoter (Fig. 2a). We transfected
plasmids encoding each of these 11 TALE-TET1 proteins into both 293 and HeLa cells
and then assayed RHOXF2 expression and promoter methylation status using quantitative
RT-PCR and high-throughput bisulfite sequencing, respectively (Supplementary Fig.
5). We successfully identified three out of six fusions that induced significant demethylation
(greater than 15%) at the −250 to +1 region in HeLa and 293 cells and another three
out of six that induced significant demethylation (greater than 15%) at the −650 to
−850 region in 293 cells. Two of the 11 TALE-TET1 proteins we tested (RH-3 and RH-4)
induced high levels of RHOXF2 mRNA expression in both the 293 and HeLa cell lines
and also demethylated proximal CpGs in the −200 to +1 region of the RHOXF2 promoter
(Figs. 2b-2d). The RH-3 fusion also binds to an additional site in the −650 to −850
region of the RHOXF2 promoter but demethylation of CpGs in this region can only be
observed in 293 cells because cytosines in this region are not methylated in HeLa
cells (Supplementary Figs. 5 and 6 and data not shown). Interestingly, we found that
even greater increases in RHOXF2 expression could be induced by combined expression
of both the RH-3 and RH-4 TALE-TET1 proteins in 293 cells (Supplementary Fig. 7).
Although we do not know the mechanism for this dramatic increase, understanding this
phenomenon will be an important focus of future studies.
To assess whether the enzymatic activity of the TET1 domain is important for the gene
activation observed with the RH-3 and RH-4 proteins, we tested variant fusions bearing
mutations (H1671Y, D1673A) known to inactivate TET1 catalytic activity.
7
We found that these catalytically inactive RH-3 and RH-4 mutants neither demethylated
their proximal CpGs nor activated RHOXF2 gene expression in either 293 or HeLa cells
(Figs. 2b, c and e). Western blots also confirmed that the observed inactivity of
these RH-3 and RH-4 mutant proteins is not due to their decreased expression in 293
cells (Supplementary Fig. 8). These results strongly suggest that activation of RHOXF2
expression is mediated by TALE-TET1-induced modification (either hydroxylation and/or
demethylation) of specific methylated CpGs in the promoter and not simply by competitive
binding of TALE-TET1 fusions with endogenous transcription factors or the presence
of a fortuitous transcriptional activation function within the fusion protein.
To further generalize these results, we next sought to demethylate CpGs in an additional
locus, the human beta-globin (HBB) gene promoter. Previous work has suggested that
four CpGs, which are differentially methylated in erythroid cells isolated from fetal
liver and adult bone marrow, may play a role in regulating HBB gene expression
17
. To test this hypothesis, we constructed ten TALE-TET1 proteins targeted to various
sites proximal to these four CpGs (Fig. 3a). Although all ten TALE-TET1 fusions (termed
HB-1 through HB-10) induced significant demethylation of CpGs near their respective
binding sites in human K562 cells (Fig. 3b and 3c), significant increases in HBB gene
expression as measured by quantitative RT-PCR were observed with only four of these
proteins (HB-3, HB-4, HB-5, and HB-6) (Fig. 3d). Of note, the three proteins (HB-4,
HB-5, and HB-6) that induced the greatest fold-activation of the promoter were the
fusions that induced the greatest demethylation of the CpG at position −266 (numbered
relative to the transcription start site; Fig. 3d). HB-4, HB-5, and HB-6 proteins
bearing the H1671Y/D1673A mutations (which inactivate TET1 catalytic domain activity)
failed to demethylate the −266 CpG and also failed to efficiently activate HBB gene
expression in K562 cells (Figs. 3e and 3f). Western blot experiments confirm that
the loss of demethylation and gene activation activities observed with the catalytically
inactive mutants of HB-5 and HB-6 is not due to decreased protein expression in K562
cells (Supplementary Fig. 9). Furthermore, time-course experiments show that both
demethylation of the −266 CpG and activated expression of HBB diminish as transfected
cells continue to be cultured (Supplementary Results and Supplementary Fig. 10), suggesting
that continued expression of TALE-TET1 is required to maintain these effects. Taken
together, our findings strongly suggest that either hydroxylation and/or demethylation
of this particular methylated CpG is required for the observed activation of HBB gene
expression.
Our results define a generalizable approach for targeting 5-methylcytosine hydroxylase
activity with subsequent cytosine demethylation to any endogenous genomic locus of
interest in living cells. While the majority of cytosines that are converted to uracil
in our bisulfite experiments are most likely to be unmethylated cytosines, it is possible
that a very small percentage of these might also represent 5-formylcytosine (5-fC)
or 5-carboxylcytosine (5-caC), further oxidation products of 5-hmC catalyzed by TET1
9
. However, given that 5-fC and 5-caC are rapidly removed from DNA via TDG–mediated
excision
18
, these oxidation products are likely to be short-lived in our cells. Therefore, we
presume that these species will be present at very low levels, as has been previously
observed in other mammalian cells
9
.
The TALE-TET1 framework described here can be easily programmed to target essentially
any DNA sequence using the simple TALE repeat code and we used this platform to induce
locus-specific demethylation at three endogenous genes (KLF4, RHOXF2, and HBB) in
three different human cell lines. Although we frequently observed the greatest degree
of demethylation within 30 bps of either end of the TALE target binding site, some
fusions also induced demethylation of CpGs approximately 150–200 bps away from the
target site, suggesting that the TET1-CD might also access regions of open chromatin
located at least one nucleosome distance away. In this study, we only examined CpGs
proximal to the TALE-TET1 binding site but it is also possible that other sites elsewhere
in the genome might also be modified due to higher-order interactions in the three-dimensional
structure of nuclear DNA.
Our success rate for making TALE-TET1 fusions capable of modifying methylated cytosines
was high but varied by target gene. For the KLF4 and HBB genes, all 12 fusions we
made (two at KLF4 and 10 at HBB) induced significant demethylation greater than 15%
at CpGs adjacent to their target binding sites; however, for the RHOXF2 promoter,
only ~50% of the fusions we made induced significant demethylation (greater than 15%).
The inability of some fusions to mediate significant demethylation might be caused
by locus-dependent effects that affect target site occupancy such as chromatin structure,
nucleosome occupancy, DNA methylation, or other parameters that could affect DNA-binding
by the TALE repeat arrays and/or the efficiency of hydroxylation by the TET1 catalytic
domain.
Our experiments provide a framework for using TALE-TET1 proteins to evaluate the functional
significance of specific CpG (and possibly non-CpG cytosine) methylation events. In
this report, we successfully identified several CpGs within the RHOXF2 promoter and
a single CpG within the HBB promoter that, when modified by either hydroxylation and/or
demethylation, are associated with an increase in gene expression. Even modest levels
of methylated CpG modification in the population of cells can, in some cases, be associated
with high levels of gene activation. We hypothesize that modification of these methylated
CpGs might allow endogenous transcription factors present in the 293, HeLa, or K562
cell lines to bind the promoter and activate expression of the endogenous gene. Although
the particular CpGs we identified in our transformed cancer cell-based experiments
may or may not be involved in normal physiologic regulation of the RHOXF2 or HBB genes,
our proof-of-principle experiments nonetheless illustrate a general strategy that
could be used in other more physiologically relevant cell types (e.g.--primordial
germ or erythroid cells) to define critical methylation events involved in the regulation
of these genes. Additionally, our TALE-TET1 fusions should provide important tools
for performing other, more detailed mechanistic studies that define how the loss of
methylation marks in turn leads to increases in promoter activity (e.g.—by enhancing
or reducing the binding of particular transcription factors).
An important and as-yet unanswered issue for future studies will be to define the
genome-wide specificities of our TALE-TET1 proteins. All of the proteins we constructed
for the RHOXF2 and HBB promoters were designed to bind 20 bp sites, sequences sufficiently
long enough to be potentially unique in the human genome. However, although previously
published in vitro SELEX experiments suggest that monomeric TALE repeat arrays are
specific for their intended target sites
19
, to our knowledge the genome-wide specificities of such proteins in human cells have
not been described. Engineered zinc finger (ZF) proteins might provide a potential
alternative to TALE repeat arrays for targeting TET1 activity and at least one published
report has suggested that monomeric six-finger proteins can be highly specific in
human cells
20
. We have also engineered multiple six-finger ZF-TET1 fusion proteins targeted to
18 bp sequences in the KLF4 and HBB genes (Supplementary Methods) and demonstrated
that these can induce targeted demethylation with efficiencies that appear to be comparable
to those induced by TALE-TET1 proteins (Supplementary Results and Supplementary Figs.
11 and 12). Regardless of which platform ultimately proves to be more specific, potential
off-target effects can be readily accounted for by constructing and testing multiple
targeted TALE-TET1 or ZF-TET1 fusion proteins for each CpG or cluster of CpGs to be
demethylated. For example, our finding that three different TALE-TET1 proteins can
all demethylate a common CpG and induce changes in HBB gene expression strongly suggests
that the observed phenotype is due to binding at the intended target sequence and
not at an off-target site elsewhere in the genome.
In addition to off-target effects resulting from unintended binding elsewhere in the
genome, it is also possible that TALE-TET1 fusions could induce demethylation that
is not dependent upon binding by the TALE repeat array portion of the protein. This
can be seen in some of our experiments with fusions targeted to an off-target site
in the EGFP reporter gene causing some level of non-specific demethylation at endogenous
loci (see, for example, Fig. 3c and 3d), presumably caused by non-DNA-bound proteins
acting from solution. Until such non-specific effects can be minimized (perhaps by
decreasing or controlling the expression level of TALE-TET1 proteins), these results
highlight the need to always perform controls with fusions targeted to other sites.
These controls will be crucial for interpreting whether phenotypic effects induced
by a particular TALE-TET1 protein depend upon the TALE repeat array-mediated sequence-specific
localization of TET1 activity
We also do not yet understand why some CpGs are more efficiently demethylated than
others by our TALE-TET1 fusions. For example, in our experiments we were able to demethylate
some CpGs in the HBB locus very efficiently (as high as 84%) whereas other CpGs in
the RHOXF2 locus were less efficiently demethylated (maximum of 42% and 25% in HeLa
and 293 cells, respectively). As noted above, this could be partly due to locus-specific
effects that affect the DNA-binding and/or hydroxylase activities of the fusions.
However, it is also possible that various factors in the cells may be actively re-methylating
CpGs and thus the extent of methylation observed may represent a steady-state between
de- and re-methylation. Our time-course results at the HBB locus (Supplementary Fig.
10) are consistent with the idea that re-methylation may be occurring in K562 cells
because demethylated CpGs appear to become re-methylated as the TALE-TET1 encoding
plasmid is lost from the cells. Delineating the parameters that affect the ultimate
efficiency of demethylation will be important to further optimize the effects of our
TALE-TET1 proteins.
The TALE-TET1 platform described here and other fusions proteins recently described
by our group
27
and others
28
represent novel and important additions to the growing toolbox of reagents for performing
targeted editing of epigenomic modifications. Previously described reagents that target
histone methyltransferases (SUV39H1 and G9A
21
) or DNA methyltransferases (bacterial enzymes
22–26
and human DNMT3a and 3b subunits
27–29
) used engineered zinc finger proteins, which can be more challenging to construct
than TALEs. Continued construction and characterization of tools to modify histones
and DNA methylation might in the future enable the stable, heritable changes in the
expression of any gene of interest. Development of such a capability in the longer-term
would enable numerous research applications as well as potential therapeutic strategies
for diseases caused by dysregulated gene expression.
Online Methods
TALE-TET1 Fusion Protein Design and Construction
The full TET1 coding sequence was synthesized as gBlocks (Integrated DNA Technologies)
and assembled by standard restriction enzyme digest and ligation to construct TET1-FL
and TET1-CD expression vectors. All TALEs were assembled using the FLASH method and
were cloned into TALE-TET1 expression vectors (pJA344C7, pJA345D4, pJA344E9 and pJA247)
containing one of four 0.5 C-terminal TALE repeats, an N-terminal nuclear localization
signal, the Δ152 TALE N-terminal domain and the +95 TALE C-terminal domain as previously
described
19,30
. In all expression plasmids used, either TET1-FL or TET1-CD was fused to the C-terminal
end of the TALE-derived DNA-binding domain via a GlyGlyGlySer linker and expression
of this fusion was driven by an EF1alpha promoter. For the Western blot experiments,
expression vectors had an additional triple-FLAG tag cloned upstream of the nuclear
localization signal. Sequences of all constructs are shown in Supplementary Fig. 13.
Cell Culture and Transfection
FlpIn-TRex HEK293 (Life Technologies) and HeLa (ATCC) cells were cultured in Advanced
DMEM supplemented with 10% FBS, 1% Glutamax, and 1% penicillin-streptomycin (Invitrogen).
K562 (ATCC) cells were maintained in RPMI supplemented with 10% FBS, 1% Glutamax,
and 1% penicillin-streptomycin. All cell lines were tested for mycoplasma every two
weeks. Plasmids encoding TALE-TET1 fusions targeted to the KLF4 and HBB loci were
transfected into K562 cells by Nucleofection. Briefly, 10 ug of TALE-TET1-encoding
plasmid (or 2, 5, 10, 20 or 50ug in the dose-response experiment) and 500 ng pmaxGFP
plasmid were Nucleofected into 1×106 K562 cells using Kit V (Lonza) and program T-016.
Control transfections used 500 ng pmaxGFP plasmid (Lonza). Fluorescent microscopy
was used to ensure consistent and high levels of transfection efficiency in all K562
experiments and FACS analysis showed ~80–90% of transfected cells are GFP+ under these
conditions. Plasmids encoding TALE-TET1 fusions targeted to human RHOXF2 were transfected
into 293 or HeLa cells using Lipofectamine LTX according to the manufacturer’s instructions
(Life Technologies). Briefly, 3.2 × 105 293 cells or 1 × 105 HeLa cells were seeded
into 12-well plates and transfected the following day with 1.2 µg TALE-TET1-encoding
plasmid, 60 ng pmaxGFP plasmid, 1 µl Plus reagent, and 3.3 µl Lipofectamine LTX. Fluorescent
microscopy was used to ensure consistent and high levels of transfection efficiency.
Cell viability in K562 cells was assayed by resuspending cells in PBS with 10% FBS
and 1 ug/ml propidium iodide and analyzing by FACS.
Genomic DNA and Total RNA isolation
Four days post-transfection, genomic DNA (gDNA) was isolated using the QIAamp DNA
Blood Mini Kit (Qiagen) according to manufacturer’s protocol. Total RNA was isolated
from cells transfected with plasmids encoding TALE-TET1 fusions targeting HBB or RHOXF2
using the PureLink RNA Mini Kit (Ambion) according to the manufacturer’s instructions.
RNA was treated with TurboDNA-Free (Ambion).
High-Throughput Bisulfite Sequencing
500 ng of genomic DNA isolated from transfected cells was bisulfite treated using
the EZ DNA methylation, EZ DNA Methylation-Gold or EZ DNA Methylation-Lightning Kit
(Zymo Research) according to the manufacturer’s instructions. All samples underwent
bisulfite conversion with an efficiency of at least 98.5% as judged by conversion
of unmethylated, non-CpG cytosines. Genomic DNA sites in KLF4, HBB, and RHOXF2 were
amplified by PCR using bisulfite-converted gDNA as a template with Kapa HiFi HotStart
Uracil+ ReadyMix (Kapa Biosystems) (for KLF4 and HBB sites) or Qiagen’s PyroMark PCR
Kit (for RHOXF2 sites). Standard Illumina adaptors were added by either ligation or
PCR and Illumina multiplex barcodes were added by PCR. For details of PCR reactions,
see Supplementary Methods. Pooled amplicons were sequenced using an Illumina MiSeq
with 150 bp paired-end reads (Dana Farber MBCF Genomics Core). For each experimental
sample assayed, we analyzed between 10,000 and 375,000 reads. Note that because RHOXF2
and RHOXF2B sequences are identical in the region examined, the bisulfite PCR analysis
does not distinguish between these two loci. For Sanger sequencing of KLF4 samples,
initial bisulfite PCR products were cloned using the TOPO Zero-Blunt cloning kit (Life
Technologies) and transformed into E. coli. Plasmid DNA was purified from the resulting
colonies and sequenced by the MGH DNA Core facility.
qRT-PCR assays
For assay of HBB gene expression, RNA was reverse transcribed using the SuperScript
III First-Strand Synthesis SuperMix and oligo-dT (Life Technologies). Quantitative
PCR was performed with Taqman Universal PCR Mastermix (Applied Biosystems) on an ABI
7500 Fast Real-Time PCR system with the following primer/probe sets: Forward HBB primer
5’-CAAGGGCACCTTTGCCACAC-3’; Reverse HBB Primer 5’-TTTGCCAAAGTGATGGGCCA-3’; HBB Taqman
Probe 5'-/56-FAM/CCTGGGCAA/ZEN/CGTGCTGGTCTGTGT/3IABkFQ/-3'; Forward β-actin (ACTB)
Primer: 5’-GGCACCCAGCACAATGAAG-3’; Reverse ACTB primer 5’-GCCGATCCACACGGAGTACT-3’;
ACTB Taqman Probe 5’-/5MAX550-Y/TCAAGATCA/ZEN/TTGCTCCTCCTGAGCGC/3IABlk_FQ/-3’.
For assay of RHOXF2 gene expression, RNA was reverse transcribed using iScript cDNA
Synthesis Kit (BioRad) according to manufacturer’s protocol. qPCR was performed with
SsoAdvanced SYBRGreen Supermix (BioRad) on an ABI StepOnePlus instrument with the
following primers: RHOXF2 Forward primer 5’-GGCAAGAAGCATGAATGTGA-3’; RHOXF2 Reverse
primer 5’-TGTCTCCTCCATTTGGCTCT-3’; M/H Actin Forward primer 5’-GTCCACACCCRCCGCCAG-3’;
M/HActin Reverse primer 5’-CCCACGATGGAGGGGAA-3’. Note that this assay does not distinguish
between RHOXF2 and RHOXF2B.
All transfections were performed in triplicate and for each biological replicate at
least three technical replicates of the qPCR assay were performed. Statistical significance
was determined by comparing experimental samples against the off-target control using
a one-sided t-test after confirming that data sets exhibited a normal distribution
as determined by a Shapiro-Wilk test for normality (p<0.05). The similarity of variance
between groups was determined using an f-test. When variance was equal between data
sets, a two-sample equal variance t-test was used and when variance was unequal, a
Welch’s t-test was used.
Accession Codes
All raw sequenced reads and BSMAP processed data files have been deposited in NCBI's
Gene Expression Omnibus and are accessible through GEO Series accession number GSE50761
(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50761) GSE50761.
Supplementary Material
1
2
Related collections
Most cited references21
- Record: found
- Abstract: found
- Article: not found
A TALE nuclease architecture for efficient genome editing.
Jeffrey Miller, Siyuan Tan, Guijuan Qiao … (2011)
- Record: found
- Abstract: found
- Article: not found
FLASH Assembly of TALENs Enables High-Throughput Genome Editing
Deepak Reyon, Shengdar Tsai, Cyd Khayter … (2012)
- Record: found
- Abstract: found
- Article: not found
Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain.
Junjie U. Guo, Yijing Su, Chun Zhong … (2011)