- Record: found
- Abstract: found
- Article: found
Estimation of data-specific constitutive exons with RNA-Seq data

Read this article at
Abstract
Background
RNA-Seq has the potential to answer many diverse and interesting questions about the inner workings of cells. Estimating changes in the overall transcription of a gene is not straightforward. Changes in overall gene transcription can easily be confounded with changes in exon usage which alter the lengths of transcripts produced by a gene. Measuring the expression of constitutive exons— exons which are consistently conserved after splicing— offers an unbiased estimation of the overall transcription of a gene.
Results
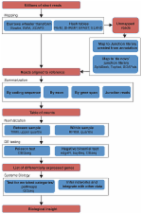
We propose a clustering-based method, exClust, for estimating the exons that are consistently conserved after splicing in a given data set. These are considered as the exons which are “constitutive” in this data. The method utilises information from both annotation and the dataset of interest. The method is implemented in an openly available R function package, sydSeq.
Related collections
Most cited references14
- Record: found
- Abstract: found
- Article: not found
Ensembl 2009
- Record: found
- Abstract: found
- Article: found
From RNA-seq reads to differential expression results

- Record: found
- Abstract: found
- Article: found