- Record: found
- Abstract: found
- Article: found
Probing Genomic Aspects of the Multi-Host Pathogen Clostridium perfringens Reveals Significant Pangenome Diversity, and a Diverse Array of Virulence Factors
Read this article at
Abstract
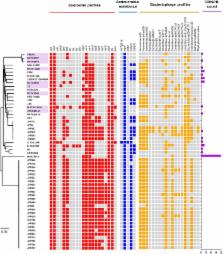
Clostridium perfringens is an important cause of animal and human infections, however information about the genetic makeup of this pathogenic bacterium is currently limited. In this study, we sought to understand and characterise the genomic variation, pangenomic diversity, and key virulence traits of 56 C. perfringens strains which included 51 public, and 5 newly sequenced and annotated genomes using Whole Genome Sequencing. Our investigation revealed that C. perfringens has an “open” pangenome comprising 11667 genes and 12.6% of core genes, identified as the most divergent single-species Gram-positive bacterial pangenome currently reported. Our computational analyses also defined C. perfringens phylogeny (16S rRNA gene) in relation to some 25 Clostridium species, with C. baratii and C. sardiniense determined to be the closest relatives. Profiling virulence-associated factors confirmed presence of well-characterised C. perfringens-associated exotoxins genes including α-toxin ( plc), enterotoxin ( cpe), and Perfringolysin O ( pfo or pfoA), although interestingly there did not appear to be a close correlation with encoded toxin type and disease phenotype. Furthermore, genomic analysis indicated significant horizontal gene transfer events as defined by presence of prophage genomes, and notably absence of CRISPR defence systems in >70% (40/56) of the strains. In relation to antimicrobial resistance mechanisms, tetracycline resistance genes ( tet) and anti-defensins genes ( mprF) were consistently detected in silico ( tet: 75%; mprF: 100%). However, pre-antibiotic era strain genomes did not encode for tet, thus implying antimicrobial selective pressures in C. perfringens evolutionary history over the past 80 years. This study provides new genomic understanding of this genetically divergent multi-host bacterium, and further expands our knowledge on this medically and veterinary important pathogen.
Related collections
Most cited references70
- Record: found
- Abstract: found
- Article: not found
Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya.
- Record: found
- Abstract: found
- Article: not found
Staphylococcus aureus Resistance to Human Defensins and Evasion of Neutrophil Killing via the Novel Virulence Factor Mprf Is Based on Modification of Membrane Lipids with l-Lysine
- Record: found
- Abstract: found
- Article: not found