- Record: found
- Abstract: found
- Article: found
Targeted Delivery of Mitochondrial Calcium Channel Regulators: The Future of Glaucoma Treatment?
discussion
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Introduction
Glaucoma is a multifactorial neurodegenerative disease affecting 64.3 million people
worldwide (Tham et al., 2014). Despite vigorous research on new treatments, those
that reduce intraocular pressure (IOP) remain the gold standard. However, their effectiveness
has been questioned as they only slow down degeneration without significantly reversing
or stopping the disease (Osborne et al., 2016b). Recent studies have, therefore, investigated
the causative roles of other processes, including glutamate toxicity, glial overactivation,
etc., (Mann et al., 2005; Chong and Martin, 2015; Lopez Sanchez et al., 2016; Vecino
et al., 2016).
Mitochondrial dysfunction is another widely studied causal process in the development
of glaucoma and has also been investigated as a potential drug target. For example,
red light therapy, manipulation of the mammalian target of rapamycin (mTOR) pathway,
and nicotinamide treatment are three recently investigated clinical therapies for
glaucoma-related mitochondrial dysfunction (Osborne et al., 2016a,b; Williams et al.,
2017). Mitochondrial activity is intimately linked to oxidative metabolism and reactive
oxygen species (ROS) formation (Schieke et al., 2006). ROS production is known to
cause retinal ganglion cell (RGC) apoptosis and subsequent vision loss. Furthermore,
while mitochondrial function is regulated by multiple pathways, calcium signaling
likely plays a key role (Vosler et al., 2008; Hurst et al., 2017). In fact, plasma
membrane calcium channel inhibitors were recently found to arrest acute axonal degeneration
and improve regeneration after optic nerve crush (Ribas et al., 2017). A different
combination of calcium permeability inhibitors also preserved optokinetic reflex following
partial optic nerve transection (Savigni et al., 2013). While the inhibitors utilized
in these studies targeted calcium channels in the plasma membrane, their effects indicate
that ROS generation and calcium signaling, which are significantly regulated by the
mitochondria, are critical during glaucoma pathogenesis.
Recently, a mitochondrial-specific drug delivery system was shown to be effective
in increasing drug concentration in mitochondria in hepatic injuries and drug-resistant
cancer cells (Yamada and Harashima, 2017; Yamada et al., 2017). However, the full
potential of this system (and other similar systems) has not been fully evaluated
with regards to calcium regulation in the diseased retina. In this opinion article,
we provide a brief discussion concerning the role of mitochondrial calcium regulation
during glaucoma pathogenesis as well as insight concerning the potential use of mitochondrial-specific
drug delivery during disease treatment. We believe that the extensive research and
overlap in the fields of glaucoma and mitochondrial disease/aging (including calcium
signaling dysfunction) ultimately lead to the therapeutic utilization of mitochondrial-specific
delivery of calcium channel regulators during glaucoma and other retinal/neurodegenerative
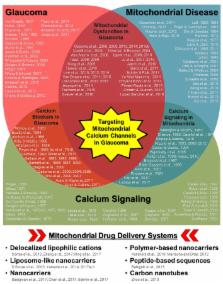
diseases (Figure 1).
Figure 1
Schematic diagram highlighting the relationships between glaucoma, mitochondrial disease/aging,
and calcium signaling along with multiple keystone studies and reviews from prominent
research groups. Some of the earliest published research concerning disease pathology
and mechanisms are listed for each respective field as well as in areas of overlap
(e.g., mitochondrial dysfunction in glaucoma, calcium channel treatment in glaucoma,
and calcium signaling in mitochondria). While it is not possible to list all of the
influential research published in each field, those listed include some of the key
historical publications, with particular emphasis on relationships with the ocular
environment or neurodegeneration when applicable. The cumulative research reported
in these publications (and those cited within) in each respective field as well as
other disease contexts has led to the development of multiple mitochondria-specific
drug delivery systems, listed in the bottom panel. Their validation in parallel with
the continued investigation of mitochondrial calcium signaling during disease pathogenesis
indicate that targeting mitochondrial calcium channels during glaucoma could be a
powerful therapeutic tool.
Glaucoma pathophysiology
Glaucoma is a two phase degenerative disease. The first phase involves a primary insult
to the RGCs (Levkovitch-Verbin et al., 2003). Confirmed risk factors/insults for glaucoma
include high IOP, ischemia, and aging. While these direct insults have classically
been investigated as the cause of glaucoma-related vision loss, recent evidence indicates
that damage to the visual cortex and/or optic nerve (i.e., distal axonopathy), which
is then propagated to the retina following stress on axonal transport systems, may
play a significant role in the initiation of the disease (Calkins and Horner, 2012;
Crish and Calkins, 2015). Ultimately, all of these insults disrupt oxygen supply and
alter retinal function. Furthermore, mitochondrial oxidative phosphorylation is significantly
less effective in the affected RGCs, and energy production depends more on glycolysis
and the tricarboxylic acid cycle. This change in energy supply causes oxidative stress
and reduced ROS consumption, leading to mitochondrial damage and further ROS accumulation
(Nguyen et al., 2011). While it has been hypothesized that RGCs can still function
normally in this reduced energy state (Osborne et al., 2016b), they are more susceptible
to secondary insults.
Secondary affronts to the RGCs can come in various forms. For example, primary insult-induced
activation of retinal microglia and astrocytes as well as altered Müller cell function
have detrimental secondary consequences related to the release of pro-inflammatory
markers as well as other cytotoxic substances, including glutamate, nitrogen oxide,
etc., in the extracellular space surrounding the RGCs. Furthermore, aged/dysfunctional
mitochondria within the RGCs can also act as secondary stressors. In aged mitochondria,
the initial increase in oxidative stress and reduced ROS consumption is amplified,
resulting in a vicious positive feedback loop involving ROS along with damage to mitochondrial
and nuclear DNA (Nguyen et al., 2011). This damage is largely irreversible as the
repair mechanisms are often impaired in aged mitochondria. ATP production in cells
with damaged mitochondria also becomes increasingly more difficult, ultimately leading
to calcium dysregulation. As calcium is a known trigger for glutamate release (Neher
and Sakaba, 2008), disrupted calcium signaling in aged mitochondria can further exacerbate
primary insult-induced glutamate toxicity. Interestingly, increased glutamate concentration
also mediates calcium influx (Wojda et al., 2008), indicating multiple points of crosstalk
between mitochondrial calcium signaling and neuronal function.
Mitochondrial calcium as a key player in glaucoma
Mitochondria have two membrane layers. At the outer membrane, calcium influx is largely
mediated through voltage-dependent anion channels (VDACs) (Cali et al., 2012; Rizzuto
et al., 2012). Some reports suggest that open-state VDACs facilitate metabolite flow
and prevent cytochrome C release, while closed-state VDACs mediate the opposite (Tan
and Colombini, 2007; Hoppe, 2010; Williams et al., 2013). Further, elevated intracellular
calcium concentrations appear to increase VDAC1 oligomerization and downstream apoptosis
(Keinan et al., 2013). This increase in oligomerization has been demonstrated to be
mediated specifically by mitochondrial, rather than cytosolic, calcium, providing
a direct link between mitochondrial calcium and apoptosis.
At the inner membrane, calcium influx from the intermembrane space into the matrix
is largely regulated by calcium-activated mitochondrial calcium uniporters (MCUs)
(Cali et al., 2012). Although MCU calcium affinity is low, their effect on calcium
concentration is significant as they mediate calcium inflow in response to the negative
membrane potential/calcium gradient created by pumping protons across the membrane
during oxidative phosphorylation. Thus, MCUs are functionally dependent on both intracellular
calcium concentration and energy demand (Tsai et al., 2017). The links between calcium
and energy are further strengthened by the calcium-dependent activation of three metabolic
enzymes, pyruvate, α-ketoglutarate, and isocitrate dehydrogenases, all of which function
in the tricarboxylic acid cycle (Cali et al., 2012). MCU function also depends on
the proximity of the mitochondria to other calcium regulating organelles, including
the endoplasmic reticulum, sarcoplasmic reticulum, and plasma membrane, which can
alter the local calcium concentration (Kirichok et al., 2004; Rizzuto et al., 2012).
Cellular calcium homeostasis, whereby nanomolar levels of free calcium are found in
the cytosol, is maintained, at least in part, via effective buffering mechanisms,
including pH and phosphate/adenosine availability. Additional intermitochondrial buffering
mechanisms involve calcium efflux via electrogenic Ca2+/3Na+ exchangers (mNCXs) and/or
electroneutral Ca2+/2H+ exchangers (mHCXs) located on the inner mitochondrial membrane.
mNCXs are the main efflux channels in excitable tissues, including RGCs, while mHCXs
are found mainly in non-excitable tissues (Hoppe, 2010). Interestingly, these efflux
systems appear to change into influx pathways during glaucoma (Wojda et al., 2008).
In aged neurons, the expression of calcium buffering proteins, including calbindin-D28k,
calretinin, and parvalbumin, is also reduced (Bu et al., 2003). Together, these changes
in efflux levels and calcium buffering protein expression significantly alter calcium
gradients, cytosolic calcium levels (Williams et al., 2013), and mitochondrial membrane
polarization (Wojda et al., 2008), resulting in altered/inefficient oxidative phosphorylation,
ROS accumulation, downstream changes in mitochondrial function, and neuronal cell
survival.
In retinal neurons, intracellular free calcium overload triggers calpain activation
which can subsequently initiate apoptotic cascades (Sharma and Rohrer, 2004; Huang
et al., 2010; Kar et al., 2010). Calpain is a calcium-dependent cysteine protease
that, once activated, cleaves pro-apoptotic B-cell lymphoma (Bcl)-2 family members
as well as apoptosis-inducing factor (AIF) and inner membrane mNCXs (Vosler et al.,
2008). Calpain activation is also related to the formation and opening of mitochondrial
permeability transition pores (mPTPs) (Cali et al., 2012; Bernardi and Di Lisa, 2015).
mPTPs are multi-protein complexes that facilitate calcium efflux. However, unlike
mNCXs, once the mPTPs are opened, the inner membrane is irreversibly permeabilized,
resulting in uncontrolled dissipation of the electrochemical gradient, ATP depletion,
ROS production, cytochrome C efflux, and mitochondrial swelling (Rasheed et al., 2017).
Notably, increased cytoplasmic cytochrome C levels not only facilitate additional
calpain activation, but the coupling of cytochrome C with apoptosis protease-activating
factor (APAF)-1 results in apoptosome formation. Apoptosomes recruit and activate
caspase-9 and downstream caspase-mediated apoptosis. Interestingly, activation of
various caspases also feeds back into the process to further activate pro-apoptotic
Bcl-2 proteins family members and increase mitochondrial permeability.
Calcium-related drug treatments in the ocular environment
Various calcium regulating therapies have been investigated for their use in treating
visual neurodegeneration (Kamel et al., 2017). Indeed, in the dorsal lateral geniculate
nucleus and superior colliculus as well as RGCs, lomerizine, a well-known plasma membrane
calcium channel blocker, has been used to manage neuronal degeneration (Ito et al.,
2010; Selt et al., 2010). Various combinations of calcium channel inhibitors, including
the L-/N-type channel blocker amlodipine, T-type channel blocker amiloride, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic
acid (AMPA) receptor blockers, and purinergic receptor blockers, also increase RGC
survival, reduce axonal degeneration, and increase axonal regeneration in both partial
optic nerve transection (Savigni et al., 2013) and crush models (Ribas et al., 2017).
However, the effects on visual function preservation were not significant.
In glaucoma and retinitis pigmentosa models, inhibition of calpain signaling has also
been demonstrated to be beneficial for RGC and photoreceptor survival, respectively.
This is not surprising as the detrimental effects of calcium overload are mediated
largely through calpain activation. Latanoprost, an ocular anti-hypertension drug,
for example, modulates its neuroprotective effects in this calpain-mediated manner
(Yamamoto et al., 2017).
Unfortunately, beyond these studies using agents against calcium signaling in the
plasma membrane, other mitochondria-specific calcium channel regulators to block detrimental
calcium changes have not been intensively studied and none have been investigated
as a glaucoma treatment option. Notably, calcium can be transported across the outer
and inner mitochondrial membranes via VDACs and MCUs, respectively, as well as during
unregulated diffusion through mPTPs, making each of these protein complexes a potential
target for calcium regulation during disease. While a number of agents have been used
to block mPTPs (Kajitani et al., 2007; Halestrap and Richardson, 2015), treatment
with these agents leaves the causative upstream changes in calcium concentration and
channel function largely unchecked. Thus, targeting VDACs and/or MCUs and avoiding
mPTP formation altogether would potentially be more advantageous. For example, an
anti-VDAC antibody has been shown to reduce cytochrome C release from mitochondria
(Madesh and Hajnóczky, 2001). Unfortunately, directly altering VDAC function in this
manner can also manipulate the transport of other essential metabolites (Camara et
al., 2017). In cancer, blocking VDACs results in apoptosis (Shoshan-Barmatz et al.,
2017), which is counterproductive to the cellular rescue required during glaucoma
treatment. Alternatively, an MCU blocker, Ru360, was demonstrated to alter ion transport
through these channels as well as block iron overload and has the advantage of having
minimal effects on other cellular functions (Sripetchwandee et al., 2013). This same
blocker was also previously shown to prevent the accumulation of mitochondrial free
calcium despite high cytosolic free calcium concentrations in post-ischaemic rat heart
cells (de Jesús García-Rivas et al., 2005). Lastly, this drug also maintains normal
oxidative phosphorylation levels and prevents mPTP opening, while other organelles
and cellular processes are unaffected, making it a drug of interest for glaucoma therapy.
Mitochondrial-specific drug delivery as a means to treat glaucoma
Organelle-specific drug targeting itself is not novel, being reviewed in multiple
excellent publications (Sakhrani and Padh, 2013; Zhang and Zhang, 2016). While mitochondrial-specific
drug targeting has not been applied to glaucoma, researchers have been actively proposing
new mitochondrial delivery/transporter systems to target this organelle in other diseases.
For example, a liposomal-based carrier was recently described that uses octaarginine
modification, electrostatic attraction, and membrane fusion to promote mitochondrial
uptake (Yamada and Harashima, 2017). This style of “MITO-porter” was then used to
deliver coenzyme Q10 in mice with hepatic ischemic/reperfusion injuries and mediated
a significant decrease in serum alanine aminotransferase (ALT) (Yamada et al., 2015).
Another study utilized a MITO-porter system to target doxorubicin to the mitochondria
of drug-resistant cancer cells, successfully destroying these cells (Yamada et al.,
2017). Other nanotechnology techniques have also been employed, including a recent
hybrid of polylactide-co-glycolide nanoparticles and mitochondria-penetrating particles
(Selmin et al., 2017; Figure 1, bottom panel). Taken together, the evidence emerging
from these investigations provides a solid foundation for the continued study of these
delivery systems in other cellular contexts.
Delivery of agents used to modulate channel function along with the expression of
other essential compounds (e.g., cytochrome C, ATP, etc.,) in concentrated amounts
directly to the mitochondria during glaucoma using these systems would allow some
of the downstream detrimental changes to be managed before vision loss. While some
current (e.g., Ru360) and future drugs targeting mitochondrial calcium channels already
innately target the mitochondria, the use of MITO-porters and similar delivery systems
would not only allow higher concentrations to be delivered, but would also avoid any
unknown effects on other organelles. Furthermore, delivery systems could also be used
to package multiple drugs/compounds together in order to have the greatest therapeutic
effect. Ultimately, these drugs would collectively reduce calcium efflux and restore
calcium homeostasis as well as prevent mPTP formation, ATP depletion, ROS production,
and cytochrome C dissipation. Doing so would prevent the second wave of apoptosis,
allowing the cells to function normally even after the initial insult. While these
drug delivery systems are not currently used as an ocular disease treatment, their
potential to transport drugs to the retina and/or optic nerve/visual cortex that will
subsequently manipulate mitochondrial function is a promising research avenue for
novel treatment development for glaucoma as well as other retinal pathologies.
Conclusions
Mitochondrial dysfunction and the associated changes in calcium homeostasis, ROS production,
and energy supply are intimately related to RGC death/dysfunction during glaucoma,
making it an attractive treatment target. Mitochondrial-targeting drug delivery systems,
which have been developed and validated in other cellular environments, could potentially
avoid these issues by packaging multiple drugs and delivering them at high concentrations
directly to the mitochondria. In discussing the recent advances in these techniques
within the context of mitochondrial calcium regulation during glaucoma for the first
time, we pose the question: Is this the future of glaucoma treatment? The relationships
highlighted in multiple keystone studies investigating glaucoma and mitochondrial
disease/aging in addition to the essential role of calcium signaling in these processes
indicate an affirmative answer. Thus, while the full potential of these systems has
yet to be fully established, we believe that mitochondrial-specific delivery of calcium
channel regulators could effectively change how glaucoma and other neurodegenerative
diseases affecting the retina are treated.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to
the work, and approved it for publication.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial
or financial relationships that could be construed as a potential conflict of interest.
Related collections
Most cited references170
- Record: found
- Abstract: found
- Article: not found
Mitochondria as sensors and regulators of calcium signalling.
Rosario Rizzuto, Diego De Stefani, Anna Raffaello … (2012)
- Record: found
- Abstract: found
- Article: not found
The impact of ocular blood flow in glaucoma.
Josef Flammer, Selim Orgül, Vital P. Costa … (2002)
- Record: found
- Abstract: found
- Article: not found