- Record: found
- Abstract: found
- Article: found
A novel de novo mosaic mutation in PHEX in a Korean patient with hypophosphatemic rickets
Read this article at
Abstract
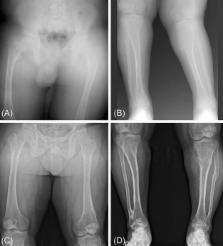
X-linked hypophosphatemic rickets is caused by loss-of-function mutations in PHEX, which encodes a phosphate-regulating endopeptidase homolog. We report a 26-year-old man with X-linked hypophosphatemic rickets who showed decreased serum phosphate accompanied by bilateral genu valgum and short stature. He had received medical treatment with vitamin D (alfacalcidol) and phosphate from the age of 3 to 20 years. He underwent surgery due to valgus deformity at the age of 14 and 15. Targeted gene panel sequencing for Mendelian genes identified a nonsense mutation in PHEX (c.589C>T; p.Gln197Ter) and a mosaic pattern where only 38% of sequence reads showed the variant allele. This mutation was not found in his mother, who had a normal phenotype. This is a case of a sporadic nonsense mutation in PHEX and up to date, this is the first case of a mosaic mutation in PHEX in Korea.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism.
- Record: found
- Abstract: found
- Article: not found
Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets.
- Record: found
- Abstract: found
- Article: not found