- Record: found
- Abstract: found
- Article: not found
Kawasaki Disease
chapter-article
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Historical Background
Kawasaki disease (KD) is one of the most common vasculitides of childhood. It has
the potential to cause severe complications, significant morbidity, and even mortality.
Expeditious treatment can largely prevent these complications, underscoring the importance
of early and accurate diagnosis. The diagnosis is based on clinical criteria (Box
35-1
), and in the absence of a diagnostic test, correct identification of KD can be as
exacting a challenge today as it has been for more than 40 years.
Box 35-1
Criteria for the Diagnosis of Kawasaki Disease
Fever for more than 5 days (4 days if treatment with intravenous immunoglobulin eradicates
fever) plus at least four of the following clinical signs not explained by another
disease process:
•
Bilateral conjunctival injection (80% to 90%)*
•
Changes in the oropharyngeal mucous membranes, including one or more of injected and/or
fissured lips, strawberry tongue, injected pharynx (80% to 90%)
•
Changes in the peripheral extremities, including erythema and/or edema of the hands
and feet (acute phase) or periungual desquamation (convalescent phase) (80%)
•
Polymorphous rash, primarily truncal; nonvesicular (>90%)
•
Cervical lymphadenopathy with at least one node >1.5 cm (50%)
This vasculitis bears the name Kawasaki disease because of the highly detailed description
of this illness in 50 children by Tomisaku Kawasaki in 1967.
1
Scattered case reports of young children who died of ruptured or thrombosed coronary
artery aneurysms have appeared in the medical literature since 1871.2, 3 A clinical
syndrome comprising most of the components of what is today recognized as KD was described
by Munro-Faure in 1959
4
and by Itoga in 1960,
5
and an even earlier fatal inflammatory vasculopathy primarily affecting young boys,
infantile polyarteritis nodosa (IPN), likely represents extreme cases of the same
disorder.
6
Definition and Diagnostic Criteria
KD is a self-limited vasculitis of unknown etiology characterized by fever, rash,
conjunctivitis, oral mucositis, extremity changes, cervical lymphadenopathy, and,
in a proportion of cases, dilation or aneurysms of the coronary and other arteries.
Criteria for the diagnosis of KD are shown in Box 35-1.
7
More recently proposed criteria include perineal rash in the criterion for changes
in the extremities, and recognition that, in the presence of fever and coronary artery
changes demonstrated by echocardiography, fewer than four criteria suffice to make
the diagnosis of KD.7, 8 Muta and colleagues
9
showed that fewer cases of KD were missed when children were included whose fevers
were abrogated with intravenous immunoglobulin (IVIG) within 5 days of the onset of
fever.
10
None of these guidelines has 100% sensitivity and specificity for the diagnosis of
KD. If a child has the characteristic clinical features and develops coronary artery
aneurysms, the diagnosis is certain. Children who do not meet the criteria may have
an incomplete or atypical form of KD (discussed later). Alternatively, some patients
who fulfill all criteria may have other conditions. In a study of patients referred
because of possible KD, Burns and colleagues
11
found that the standard clinical diagnostic criteria for KD were fulfilled in 18 (46%)
of 39 patients in whom other diagnoses were established. Furthermore, Benseler et al.
found that in a consecutive series of children diagnosed with KD, up to one third
had concurrent, identifiable infections,
12
including Group A streptococcal tonsillitis, viral illnesses, pneumonia, and gastroenteritis.
More concerning from the perspective of trying to prevent disease sequelae is that
many children who develop coronary artery aneurysms never meet criteria for KD.
13
Witt et al. found in a single center study of 127 patients treated for KD that 36%
did not meet the criteria for KD. Furthermore, the KD cases that did not meet the
criteria for diagnosis had a significantly higher proportion of coronary artery abnormalities
as compared to those cases that did meet criteria (20% vs. 7%, P < 0.05).
13
Sudo et al. utilized epidemiological data from the twentieth nationwide survey of
KD in Japan and found that the prevalence of coronary artery lesions 1 month after
disease onset tended to be higher in the cases with one or two principal criteria
as compared with those cases that had five to six criteria (7.4% vs. 2.5% P < 0.05).
14
Consistent with this, a recent meta-analysis of over 20 studies of patients with KD
found that incomplete KD is a risk factor for coronary artery abnormalities.
15
The youngest patients are the least likely to meet the classic criteria, and unfortunately
they also have the highest risk of developing coronary artery abnormalities.
16
Rosenfeld et al. found that up to 60% of children younger than 12 months old developed
aneurysms in one series.
17
For this reason, the diagnosis of KD should be considered in any infant with prolonged,
unexplained fever, and there should be a low threshold for performing echocardiography
in this age group. Conversely, in older children, treating KD is seldom an emergency,
especially when patients present symptoms after only 5 or 6 days of fever. Observation
of children older than 6 months who do not fulfill criteria may be the best course
of action. The mean duration of fever in children with untreated KD is 12 days,
18
much longer than typical viral illnesses, so the persistence of fever or development
of additional signs of KD can be an indicator for treatment.
Epidemiology
Although worldwide in distribution, the incidence of KD is highest in Japan and is
steadily increasing over time, although the reasons behind this increase are unclear.
In 2010, the annual incidence rate recorded in Japan was 239.6 per 100,000 children
aged up to 4 years, which is higher than during any of the three epidemics of KD in
Japan in 1979, 1982, and 1986. Children of Japanese descent who reside outside Japan
also face a higher risk of KD than do Caucasian children.
19
The incidence is increasing in South Korea as well, with the second highest worldwide
incidence of 134.4/100,000 children younger than 5 years of age in 2011.
20
Rates in Taiwan
21
and China
22
are also high.
The epidemiology of KD in the United States differs from countries in Asia in that
there are fewer cases (20.8/100,000 children <5 years in 2006) and the incidence does
not seem to be rising over time.
23
As assessed by hospital admissions in the U.S., children of Asian or Pacific Island
ancestry have the highest incidence (30.3/100,000 <5 years). The incidence was intermediate
for African Americans (17.5/100,00 <5 years) or children of Hispanic origin (15.7/100,000
<5 years), and lowest for whites (12/100,000 <5 years).
23
In one large area of Great Britain, the annual incidence rate was 5.5 cases per 100,000
for children younger than 5 years old; the incidence for children of Asian ancestry
was more than double that for Caucasian and African or Afro-Caribbean children.
24
KD is an illness of early childhood. Seventy-seven percent of affected patients are
younger than 5 years old, with an average age of approximately 3 years,
23
although there are reports of KD occurring in older children
25
and adults.26, 27, 28 KD is more common in boys than in girls (male-to-female ratio
of 1.36:1 to 1.62:1).29, 30
In Japan, the highest incidence occurs between 6 and 11 months old.
31
In North America, the peak age at onset of KD is between 2 and 3 years old. In a recent
Australian study of the years 2000 through 2009, the mean age of diagnosis was 4.2
years.
32
The reasons for the geographic differences in age at onset are unclear.
33
Several reports document a seasonal incidence of KD.33, 34, 35 Burns et al. established
a global seasonal pattern of KD with a peak in disease in January through March in
the extratropical Northern Hemisphere.
35
In Japan, the disease occurs most frequently in winter and spring months, with a nadir
in October.
31
In a study from Taiwan, the highest incidence was in the summer.
36
In North America, cases have tended to occur between November and May.
37
There were no seasonal variations in the incidence of KD in a study from western Australia.
32
Although epidemics of KD were documented in Japan up to 1987, none has occurred since
then.
38
In Japan, siblings of affected children have a risk of contracting KD that is approximately
10 times higher than the risk in the general population,
39
but cases among children sharing the same home in other countries are uncommon.
34
Dergun and colleagues
40
reported 18 families in the United States with 24 affected members, including 9 sibling
pairs. Second and even third attacks have been reported in a range of 1.5% to 3% of
cases.31, 36
Etiology and Pathogenesis
The cause of KD remains unknown. Many of its epidemiological and clinical manifestations
suggest an infectious origin. If an infectious agent does indeed cause KD, the putative
organism would appear to be of very low communicability, or predominantly responsible
for subclinical infections. Repeated attempts to identify a particular infectious
trigger have been unsuccessful.
41
A predominance of immunoglobulin A (IgA)-secreting plasma cells in the blood vessel
walls of children with fatal KD has suggested to Rowley and colleagues that an organism
that gained entry through mucosal surfaces underlies the disease.
42
No single pathogen is regularly demonstrable, although associations with Epstein–Barr
virus (EBV),
43
rotavirus,
44
other viruses,45, 46 and with bacteria47, 48 have been reported. An association with
a coronavirus
49
was not confirmed.
50
It is nonetheless a possibility that the vascular injury in KD may be the result of
a direct cell-mediated attack on endothelial cells that are infected with an unidentified
pathogen.
51
Other investigators have proposed that the vasculitis in KD is caused by either conventional
antigens or superantigens that trigger an immune response to endothelial cells, rather
than by direct infection of the vessels.
52
Superantigens are produced by several bacteria, notably certain strains of Staphylococcus
and Streptococcus, and are capable of stimulating large numbers of T cells in an antigen-nonspecific
manner by interaction with the β chain of the T-cell receptor. Overrepresentation
of T cells bearing Vβ2 among lymphocytes in coronary artery aneurysms, intestinal
mucosa,53, 54 and peripheral blood
55
from patients with KD supports the hypothesized role of superantigens in the pathogenesis.
A variety of additional circumstantial evidence52, 56, 57, 58 and a murine model of
Lactobacillus casei–induced vasculitis lend credence to this theory.
59
Further, children with KD have unique reactions to mycobacterial antigens,60, 61,
62 which may also function as superantigens, including recall reactions at the site
of a previous bacillus Calmette–Guérin immunization.
61
Nonetheless, the only human illness definitively ascribed to superantigens is toxic
shock syndrome, and different groups have published conflicting evidence regarding
the isolation of superantigen-producing organisms, the detection of superantigen proteins,
and the presence of an immunological signature of superantigen activity in patients
with KD.55, 63, 64
Additional clues to the cause of KD may come from humoral factors, including antiendothelial
cell antibodies, circulating immune complexes,
65
and antineutrophil cytoplasm antibodies (ANCAs) that are demonstrated by some researchers
in a proportion of patients,65, 66 but not by others.67, 68 For example, findings
from a murine model indicated that B cells may not be necessary for coronary arteritis.
69
However, genome-wide association studies have implicated single nucleotide polymorphisms
(SNPs) in the B lymphoid tyrosine kinase and CD40 genes as evidence that B cells may
play a pathogenic role in KD.70, 71
Recent studies have demonstrated that regulation of T-cell activation may determine
susceptibility to and severity of KD.
59
Onouchi et al. identified a functional SNP in the inositol 1,4,5-triphosphate 3-kinase
C (IPTKC) gene on chromosome 19 through linkage disequilibrium mapping. ITPKC acts
as a negative regulator for T-cell activation, and the SNP was associated with risk
for developing KD and coronary artery lesions.
72
Supportive evidence for the role of T cells in KD has recently been provided via an
animal model of KD, in which mice are injected intraperitoneally with Lactobacillus
casei cell wall extract. T-cell costimulation was identified as a critical regulator
of susceptibility to and severity of coronary arteritis in this model.
59
In the absence of confirmed evidence of a single etiological agent, a reasonable working
hypothesis is that KD represents a stereotyped, pathological immune response to one
or a variety of environmental and/or infectious triggers. It may be a form of reactive
vasculopathy, like polyarteritis nodosa or Henoch–Schönlein purpura, with an inflammatory
response developing in sterile tissues as a result of immune activation.
73
Presumably, certain individuals are predisposed by virtue of their genetic constitution.
The strong predilection for childhood onset may reflect the presence of developmental
antigens that are targets for the inflammatory response only early in life, subtle
maturational defects in immune responsiveness,
74
or the timing of exposure to environmental triggers.
Genetic Background
In Japan, approximately 1% of patients with KD have a history of an affected sibling,
75
and concordance for KD was 13.3% in dizygotic twins and 14.1% in monozygotic twins.
76
Similarly, in Japan there is a significantly increased frequency of a history of KD
in the parents of children with the disease.
77
These observations indicate that there is a genetic predisposition to this disease,
although the fact that affected twin pairs became ill within 2 weeks of each other
also suggests an important role for an environmental agents.
The exact genetic factors that may underlie the disorder are unknown. Reported genetic
associations have been reviewed by Hata and Onouchi.
78
Candidate genes include those at the histocompatibility locus and those for other
proteins involved in immunoregulation. Human leukocyte antigen (HLA) genes for B5,
B44, Bw51, DR3, and DRB3*0301 have been associated with KD in Caucasians; Bw54, Bw15,
and Bw35 in the Japanese; and Bw51 in Israelis.
79
Onouchi and colleagues
80
concluded that HLA polymorphisms contributed little to the pathogenesis of KD. However,
a recent study identified an SNP that achieved genome-wide significance in the HLA-DQB2-HLA-DOB
locus.
81
There has been no reported association of any HLA antigen with the risk of coronary
artery disease.
82
Polymorphisms of the tumor necrosis factor (TNF)-α gene (TNF),
83
the IL 18 gene,
84
the HLA E gene,
85
and the gene for angiotensin-converting enzyme86, 87 have also been associated with
KD, but their pathogenic significance is disputed. A separate report implicated polymorphisms
of the mannose-binding lectin (MBL) in the pathogenesis of KD.
88
MBL binds to n-acetyl glucosamine and mannose present on the surface of many microbes.
This interaction results in activation of complement (C3) independent of antibody.
Levels of MBL are determined by polymorphisms of the MBL 2 gene and its promoters.
Higher expression of MBL is associated with lower incidence of coronary artery lesions
in patients under 1 year old, but it has the opposite effect in older patients. This
apparent paradox is congruent with the belief that MBL is important in protecting
the very young child from infectious diseases.
88
The gene controlling expression of inositol 1,4,5-triphosphate 3-kinase (ITPKC) has
been identified as a susceptibility gene not only for KD, but also for coronary artery
disease.
72
This enzyme is strongly expressed by peripheral blood mononuclear cells and has an
important role in inflammation by decreasing interleukin (IL)-2 expression. The functional
polymorphism ITPKC 3 is significantly increased in patients with KD and coronary artery
disease in Japanese and American populations.
89
An SNP in the caspase-3 gene on chromosome 4 has also been associated with risk for
IVIG resistance and coronary artery abnormalities,
90
which may be due to its role in apoptosis. An SNP in the FAM167A-BLK gene is a recently
identified locus that is significantly associated with a risk for KD.70, 81 It is
of interest, as previous studies have reported an association between SNPs in this
region and autoimmune diseases, including rheumatoid arthritis
91
and systemic lupus erythematosus.92, 93 There have also been reports of associations
of functional SNPs in immunoglobulin receptor pathways81, 94 and in the gene for CD40.71,
81
Clinical Manifestations
Disease Course
The course of untreated KD may be divided into three phases (Fig. 35-1
): An acute, febrile period lasting for 10 to 14 days is followed by a subacute phase
of approximately 2 to 4 weeks. This ends with a return to normal of the platelet count
and erythrocyte sedimentation rate (ESR). The subsequent convalescent or recovery
period lasts months to years, during which time vessels undergo healing, remodeling,
and scarring.
FIGURE 35-1
Kawasaki disease can be viewed as an illness with acute, subacute, and recovery phases.
The temporal characteristics outlined here are typical of the course of the disease.
(Adapted from Kawasaki T, Acute febrile mucocutaneous syndrome with lymphoid involvement
with specific desquamation of the fingers and toes in children, Arerugi 16 (3) (1967)
178–222. [Article in Japanese].)
Acute Febrile Phase
The onset of fever in KD is characteristically abrupt, sometimes preceded by symptoms
of an upper respiratory or gastrointestinal illness. Baker and colleagues
95
studied the symptoms in the 10 days prior to diagnosis of KD in 198 patients. They
reported that irritability occurred in 50%, vomiting in 44%, decreased food intake
in 37%, diarrhea in 26%, and abdominal pain in 18%. Cough was reported in 28%, and
19% had rhinorrhea. In addition, 19% reported weakness, and 15% reported arthralgia
or arthritis. Perineal desquamation may be another early sign of KD.
96
Over the next 3 to 4 days, cervical adenitis, conjunctivitis, changes in the buccal
and oral mucosa, a pleomorphic rash, and erythema and edema in the hands and feet
develop. The manifestations occur in no particular order and can fluctuate in the
first 7 to 10 days of illness. Accordingly, a thorough medical history is important
for identifying subtle or transient manifestations of KD. Untreated, the clinical
signs of KD subside after an average of 12 days. If myocarditis occurs, it often does
so early and may be manifested by tachycardia, an S3 gallop, and perhaps signs of
congestive heart failure.
97
Pericarditis, abdominal pain, ascites, and hydrops of the gallbladder also may occur
at this time.
Subacute Phase
After the acute phase, the child may be entirely asymptomatic if given IVIG. During
this period, desquamation of the skin and specifically, periungual desquamation of
the digits,
98
may be the only clinically apparent residual feature. One in 13 children may develop
arthritis of one or several joints during the late acute and subacute phases.
99
Coronary artery aneurysms most commonly first develop during the subacute phase or
occasionally earlier, but rarely later in children treated with IVIG. The irritability
that can be quite prominent in the acute phase diminishes and resolves completely
during the subacute phase.
Convalescent Phase
Most children are asymptomatic during the convalescent phase. The acute phase response
has usually returned to normal, unless there are complications. Horizontal ridging
of the nails (Beau lines), characteristic of many acute inflammatory conditions, may
appear during this period.
Clinical Characteristics of the Classification Criteria
Fever
Fever, often exceeding 40°C (104°F), is the hallmark of KD. The fever is typically
persistent and minimally responsive to antipyretic agents, tending to remain above
38.5°C (101.3°F) during most of the acute phase of the illness. It reflects elevated
levels of TNF-α and IL-1, which are thought to mediate the underlying vascular inflammation.
100
The diagnosis must be suspect in the absence of fever, though brief temperature spikes
may be missed by fatigued or inexperienced parents.
Conjunctivitis
Bilateral, nonexudative bulbar conjunctivitis occurs in more than 85% of patients
with KD. Conjunctival injection typically spares the limbus, which is the zone immediately
around the cornea. However, the characteristic of limbal sparing is not required for
conjunctival erythema to qualify as a diagnostic criterion. Inflammation of the palpebral
conjunctiva is not prominent. Purulent discharge is especially unusual
101
and suggests an alternative diagnosis.
Other ocular abnormalities also may occur, although they are not part of the diagnostic
criteria. During the first week of illness, about three fourths of children are photophobic,
a consequence of anterior uveitis,
102
which peaks between 5 and 8 days of illness and is more common in children over 2
years old. Ocular inflammation usually resolves without specific therapy or sequelae.
In exceptional instances, there may be posterior synechiae, scleral
100
or conjunctival
103
scarring, changes in the retina and vitreous,
104
or even blindness.
105
Changes in the Lips and Oral Mucosa
Swollen, vertically cracked red lips and a strawberry tongue are characteristic of
KD; the latter is caused by sloughing of filiform papillae and prominence of the large
hyperemic fungiform papillae (Fig. 35-2
). Diffuse erythema of the oropharynx is also seen. Vesicles, ulcers, or tonsillar
exudate suggest a viral or bacterial infection rather than KD.
FIGURE 35-2
A, The intense reddening, swelling, and vertical cracking of the lips are characteristic
of Kawasaki disease (KD). B, The strawberry tongue of acute KD with hypertrophied
papillae on an erythematous base and the peeling of the facial skin.
Exanthem
The cutaneous manifestations of KD are protean. Although the rash usually begins on
the trunk, there is often a perineal confluence during the first days of the illness,
followed by desquamation in the diaper area by day 6 in many cases.
96
Macular, morbilliform, or targetoid lesions of the trunk and extremities are most
characteristic. The rash is seldom pruritic, and vesicular or bullous lesions are
rare (Fig. 35-3
). Psoriasis has been reported in several children with KD.
106
FIGURE 35-3
The nonspecific polymorphous rash is seen on the face arms and chest of this 2-year-old
boy with acute Kawasaki disease.
Lymphadenopathy
Anterior cervical lymphadenopathy occurs during the acute phase of the disease, is
usually unilateral, and may appear to involve only a single node. However, ultrasonography
or computed tomographic imaging of the neck typically reveals grapelike clusters of
enlarged nodes similar to those seen in EBV infections rather than the isolated adenopathy
typical of bacterial adenitis.
107
Occasionally, a node enlarges rapidly and may be mistaken for infectious lymphadenitis.
After 3 or 4 days, it usually shrinks with or without specific therapy. Diffuse lymphadenopathy
and splenomegaly are not typical of KD and should raise suspicions of a viral illness.
Extremity Changes
Indurated edema of the dorsum of the hands and feet and a diffuse red-purple erythema
of the palms and soles occur early and last for 1 to 3 days. Sheetlike desquamation
typically occurs 10 days or more after the start of the fever. It characteristically
begins at the tips of the fingers followed by the toes, just below the distal edge
of the nails (Fig. 35-4
). Flaky desquamation may occur elsewhere, but acral skin peeling usually occurs late
in the course of KD and may be absent or inapparent.
98
Consequently, it is more useful for retrospective confirmation of the diagnosis rather
than for making therapeutic decisions.
FIGURE 35-4
A, Desquamation of the skin of the tips of the thumb and finger seen during the subacute
phase of Kawasaki disease. B, Desquamation of the skin of the hand occurs later in
the subacute and early recovery phase of the disease. In many children, the degree
of desquamation is much less than is depicted here.
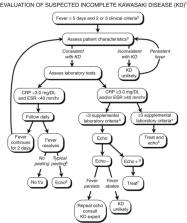
Incomplete Kawasaki Disease
Signs and symptoms in children who do not meet criteria for KD tend to parallel those
of children who fulfill the diagnostic criteria.
108
Incomplete KD is seen more frequently in young infants and older children, which is
a critical observation as these groups of children are also at higher risk for coronary
artery lesions.25, 31, 109, 110 A particularly high level of suspicion is needed in
infants younger than 1 year old. In a retrospective review of 45 cases of KD, 5 (45%)
of 11 infants had incomplete disease, compared with 4 (12%) of 33 older children.
111
In this study, coronary artery complications occurred in three older children (9%)
but in seven infants (64%), including all five with incomplete disease.
111
In view of these data, the American Heart Association (AHA) has suggested additional
markers for identification of children who do not meet the classic criteria for KD
but who might nonetheless be at increased risk for developing coronary artery aneurysms
(Fig. 35-5
).
7
Reports suggest that the algorithm recommended by the AHA committee performs well
in reducing the number of children who are not treated for KD but who ultimately develop
aneurysms.
112
FIGURE 35-5
Evaluation of suspected incomplete Kawasaki disease. 1In the absence of gold standard
for diagnosis, this algorithm cannot be evidence based but rather represents the informed
opinion of the expert committee. Consultation with an expert should be sought anytime
assistance is needed. 2Infants 6 months old on day 7 of fever without other explanation
should undergo laboratory testing and, if evidence of systemic inflammation is found,
an echocardiogram, even if the infants have no clinical criteria. 3Characteristics
suggesting disease other than Kawasaki disease include exudative conjunctivitis, exudative
pharyngitis, discrete intraoral lesions, bullous or vesicular rash, or generalized
adenopathy. Consider alternative diagnoses. 4Supplemental laboratory criteria include
albumin 3.0 g/dL, anemia for age, elevation of alanine aminotransferase, platelets
after 7 d 450 000/mm3, white blood cell count 15 000/mm3, and urine 10 white blood
cells/high-power field. 5Can treat before performing echocardiogram. 6Echocardiogram
is considered positive for purposes of this algorithm if any of 3 conditions are met:
z score of LAD or RCA 2.5, coronary arteries meet Japanese Ministry of Health criteria
for aneurysms, or 3 other suggestive features exist, including perivascular brightness,
lack of tapering, decreased LV function, mitral regurgitation, pericardial effusion,
or z scores in LAD or RCA of 2–2.5. 7If the echocardiogram is positive, treatment
should be given to children within 10 d of fever onset and those beyond day 10 with
clinical and laboratory signs (CRP, ESR) of ongoing inflammation. 8Typical peeling
begins under nail bed of fingers and then toes.
(American Heart Association, Diagnosis, treatment, and long-term management of Kawasaki
disease. Circulation 110 (2004) 2747–2771.)
Other Clinical Manifestations of Kawasaki Disease
Table 35-1
shows other clinical manifestations of KD.
TABLE 35-1
Manifestations of Kawasaki Disease
ORGAN SYSTEM
COMMON
UNCOMMON
FINDING SUGGESTS ALTERNATE DIAGNOSIS
Skin
Targetoid, urticarial, morbilliform rashes, livedo reticularis
Psoriasiform rash
Pustular, vesicular rashes
Lungs
Pleural effusion
Nodules, interstitial infiltrates
Urinary tract
Urethritis, pyuria
Hematuria, proteinuria, orchitis
Nervous system
Irritability, lethargy, anterior uveitis, sensorineural hearing loss
Seizure, stroke, cranial nerve palsy
Gastrointestinal system
Diarrhea, vomiting, hydrops of gallbladder, hepatomegaly
Intestinal hemorrhage, ruptured viscus
Hematological system
Anemia, thrombocytosis, leukocytosis
Thrombocytopenia, consumptive coagulopathy, hemophagocytic syndrome
Lymphocytosis*
Reticuloendothelial system
Anterior cervical lymphadenopathy
Posterior cervical, axillary lymphadenopathy
Diffuse lymphadenopathy, splenomegaly
Mucosa
Mucositis, glossitis, conjunctivitis
Discrete oral lesions, exudative conjunctivitis
Musculoskeletal system
Extremity edema, arthritis
Raynaud phenomenon
Cardiac system
Tachycardia, gallop rhythm, myocarditis, pericarditis
Coronary artery aneurysm, aortic root dilation, valvulitis
*
Except during the convalescent phase.
Cardiovascular Disease
At onset, there is nearly always tachycardia, typically commensurate with the degree
of fever. Early myocarditis occurs in at least one third
113
to half
114
of patients, and pericarditis also may occur.
115
Myocardial involvement often leads to decreased contractility, commonly manifested
by an S3 gallop that may become more prominent with hydration. Tachycardia out of
proportion to the fever is also found in children with significant myocarditis. Such
children may be misdiagnosed with viral myocarditis. In more severe cases, myocardial
involvement may progress to dysrhythmias and signs of congestive heart failure.
116
Children with prominent KD-associated myocarditis tend to respond briskly to treatment
with IVIG, and long-term abnormalities of cardiac contractility are very uncommon
in children treated appropriately during the acute phase of KD.
117
Tacke et al. performed cardiac magnetic resonance (MR) imaging in patients and controls
at a median of 11.6 years following diagnosis of KD and found that only those with
severe coronary artery pathology had evidence of cardiac dysfunction.
118
Recently, there has been increasing awareness of a shocklike syndrome that can occur
with KD (KD shock syndrome [KDSS]).
119
Kanegaye and colleagues identified 13 children with systolic hypotension, which is
a 20% or greater decrease in baseline systolic blood pressure, or clinical signs of
poor perfusion from a cohort of 187 consecutive patients with KD at a single center.
119
They found that a third of the children with KDSS had impaired left ventricular systolic
function and nearly two thirds had coronary artery abnormalities. IVIG resistance
was also seen more commonly in the patients with KDSS. A Taiwanese group performed
a case control study of 9 patients with KDSS compared with 27 season-matched controls
and also found that the cases had a higher risk for coronary artery dilation.
120
The most significant and characteristic complication of KD, the development of coronary
artery aneurysms in up to 25% of untreated patients, makes KD the leading cause of
acquired heart disease among children in the developed world (Figs. 35-6
and 35-7
). Frank aneurysms are unusual early in the course of disease, but the lack of tapering
seen on echocardiograms is typical, and coronary artery dimensions may be increased
in the first 5 weeks after the disease first manifests. Interestingly, Muniz et al.
compared coronary artery dimensions of febrile patients with non-KD illnesses to KD
patients, and found that the non-KD febrile controls exhibited enlarged coronary artery
dimensions, although not to the same degree as the KD cases.
121
Similarly, Bratincsak found that no febrile controls had coronary artery diameters
more than 2.5 standard deviations above the mean for age, size, and gender, a diameter
typically reached by more than 10% of children with KD.
122
FIGURE 35-6
Echocardiographic demonstration of aneurysms of three coronary arteries in a child
with Kawasaki disease. A, Aneurysms; CIRC, circumflex; LAD, left anterior descending
coronary artery; RVOT, right ventricular outflow tract.
(Courtesy Dr. Dennis Crowley.)
FIGURE 35-7
A, Angiography of the coronary vessels in a 7-month-old boy with Kawasaki disease
shows a huge aneurysmal dilation of the right coronary artery (arrow). B, Aneurysm
of the left coronary artery in a 3-year-old girl with Kawasaki disease (arrow).
(Courtesy Dr. Zuidi Lababidi.)
The Japanese Ministry of Health (JPH) criteria123, 124 use angiography or echocardiography
to define coronary arteries as abnormal if the internal lumen diameter is greater
than 3 mm in children younger than 5 years old or greater than 4 mm in children at
least 5 years old. In addition, vessels are considered aneurysmal if the internal
diameter of a segment measures at least 1.5 times that of an adjacent segment or the
coronary artery lumen is clearly irregular. Aneurysms can be defined as small (an
internal diameter of <5 mm), medium (an internal diameter of 5 to 8 mm) or giant (an
internal diameter >8 mm) per the JPH.
Although coronary artery dimensions in normal children have been shown to increase
linearly with body surface area (BSA) or length,
125
the JPH criteria are not based on body size. Evaluation of coronary arteries in KD
using age-, size-, and sex-adjusted indices (z scores) suggests that the incidence
of abnormalities is higher than was generally recognized.
126
Among patients classified as having normal coronary arteries by the JPH criteria,
27% had at least one BSA-adjusted coronary artery dimension more than 2 standard deviations
above the mean. Of note, z scores are available only for the left main coronary artery,
the left anterior descending artery, and the right coronary artery. Other coronary
vessels can be assessed using the JPH criteria. Even children whose vessel dimensions
are within the “normal” range may demonstrate a decrease in coronary artery diameter
as they convalesce from KD.
127
Some experts think that a z-score-based system of classifying aneurysms may be more
discriminating, with a giant aneurysm defined as a z score of 10 or higher.
Coronary aneurysms may cause morbidity early in the course due to rupture or thrombosis,
resulting in sudden death or myocardial infarction.
128
Development of de novo coronary artery abnormalities more than 2 weeks after the end
of the acute illness is unusual.
Although involvement of the coronary arteries is the most characteristic manifestation
of the vasculitis of KD, other medium-sized muscular arteries also may be involved.
Aneurysms of brachial and femoral arteries may be palpable clinically or demonstrable
angiographically (Fig. 35-8
). In severe cases, peripheral arterial obstruction may lead to ischemia and gangrene.
Visceral arteries are usually spared, although there are reports of gastrointestinal
obstruction
129
and acute abdominal catastrophe
130
occurring because of vasculitis. Such complications generally arise in children with
other signs of severe vasculitis, including aneurysms in coronary and peripheral arteries.
FIGURE 35-8
Angiographic study of a 2-year-old boy with severe Kawasaki disease resulting in multiple
aneurysms of the coronary, axillary, iliac, and femoral arteries. The study revealed
large aneurysms of the aorta and iliac arteries (A) and the femoral arteries (B;
arrows). Aneurysms that were palpable in the axilla and groin in this patient later
resolved.
(Courtesy Dr. G. Culham.)
Central Nervous System Complications
One of the most consistent clinical observations of children with KD, particularly
in infants and very young children, is their extreme irritability. This probably represents
the effect of aseptic meningitis and associated headache.
131
Cerebrovascular accident132, 133 and facial nerve paralysis
134
have also been reported.
Musculoskeletal Disease
Arthritis was observed by Gong and colleagues
99
in 7.5% of 414 children with KD. Arthritis was oligoarticular in 55% and polyarticular
in 45%. Joints most commonly affected were (in order of decreasing frequency) knee,
ankle, wrist, elbow, and hip. Joint pain was often severe, but responded to IVIG and
high-dose aspirin in most instances. It may occur at any time during the disease course
but has been described most commonly during the recovery phase. Arthritis in KD ultimately
resolves, leaving no residua.
Respiratory Tract Disease
Cough, coryza, hoarseness, and otitis media frequently occur early in the course of
the disease, and suggest a viral upper respiratory tract infection. Approximately
one third of children have some degree of sensorineural hearing loss when tested within
30 days of fever onset. Salicylate toxicity may be responsible for transient cases,
but sensorineural hearing loss of unclear etiology rarely may persist after aspirin
is discontinued.135, 136, 137
Gastrointestinal Tract Disease and Other Abnormalities
Abdominal pain is common, and approximately one fourth of children with KD have profuse,
watery diarrhea during the acute febrile period. Abdominal distention may mimic mesenteric
vasculitis or intussusception, and children with KD can present with an acute surgical
abdomen, although this is rare.
130
Segmental bowel wall thickening has been described in children with KD and abdominal
pain, presumably reflecting visceral arteritis.
138
The relatively common occurrence of hydrops of the gallbladder demonstrated by ultrasonography
139
may aid in the diagnosis of incomplete KD. Occasionally, the gallbladder becomes large
enough to be seen as a bulge in the anterior abdominal wall. The specificity of gallbladder
distension is limited, however, and a dilated, engorged gallbladder may be seen in
cases of streptococcal and staphylococcal infections, among other mimics of KD. Hepatosplenomegaly
may occur in the absence of heart disease, or it may reflect cardiac failure.
Genitourinary Tract Involvement
Kidney and genitourinary tract involvement is uncommon but reported in KD.
140
A study of 50 children with KD from Taiwan
141
revealed hematuria (>5 red blood cells per high-power field [RBC/HPF]) in 6 patients,
proteinuria (>100 mg/dL) in 5, and leukocyturia (>10 white blood cells per high-power
field [WBC/HPF]) in 19. Renal ultrasonography was abnormal in five patients, and dimercaptosuccinic
acid single photon emission computed tomography (DMSA SPECT) revealed inflammatory
lesions in 26 children. Although renal function remained normal, scarring was demonstrated
in 46% on repeated DMSA SPECT. Sterile pyuria is one of the supplemental laboratory
criteria in the algorithm for suspected incomplete KD. Burns et al. compared pyuria
in KD cases versus febrile controls without urinary tract infections. They found that
pyuria was neither sensitive nor specific for KD, but that the magnitude of pyuria
in KD was significantly higher than in febrile controls (42 WBC/µL vs. 12 WBC/µL).
142
Scrotal pain and swelling due to testicular inflammation are characteristic of pediatric
vasculitides, including Henoch–Schönlein purpura, polyarteritis nodosa, and KD.
143
Meatitis and dysuria also occur frequently during the acute phase of KD, and priapism
has been described.
144
Hemolytic-uremic syndrome, immune complex–mediated glomerulonephritis, and acute interstitial
nephritis have each been reported in a few cases.145, 146 Acute renal failure is a
rare complication most commonly ascribed to complications of treatment with certain
preparations of IVIG.
147
Differential Diagnosis
The differential diagnosis of KD includes viral and bacterial infections, toxin-mediated
diseases, systemic-onset juvenile idiopathic arthritis and Stevens–Johnson syndrome
(Box 35-2
). Viral illnesses such as measles (especially when atypical or occurring after vaccination),
EBV, and adenovirus infections share many of the signs of mucocutaneous involvement,
but they typically have less evidence of systemic inflammation and generally lack
the extremity changes of KD. Toxin-mediated illnesses, especially scarlet fever, staphylococcal
scalded skin syndrome, and toxic shock syndrome lack the ocular and articular involvement
typical of KD. Drug reactions, such as those in Stevens–Johnson syndrome or serum
sickness, may mimic KD but have subtle differences in the ocular and mucosal manifestations.
In particularly severe or prolonged KD, the possibility of a chronic vasculitis such
as polyarteritis nodosa
148
must be considered carefully. A lack of renal involvement and presence of mucocutaneous
changes favor the diagnosis of KD over polyarteritis nodosa.
Box 35-2
Differential Diagnosis of Kawasaki Disease
Infectious Conditions
Adenovirus
Measles
Parvovirus
Human herpesviruses (HHV) (e.g., herpes simplex virus, cytomegalovirus, HHV-6, HHV-7)
Rocky Mountain spotted fever
Leptospirosis
Streptococci
Staphylococci
Immune System Reactions
Stevens–Johnson syndrome
Toxin-mediated diseases (toxic shock syndrome)
Serum sickness
Rheumatic Diseases
Systemic-onset juvenile idiopathic arthritis
Polyarteritis nodosa
Pathology
The signs and symptoms of KD are due to a systemic necrotizing vasculitis with fibrinoid
necrosis of the medium-sized muscular arteries; the coronary arteries are the predominant
sites of involvement.
149
Disruption of the lamina elastica is characteristic of the aneurysms. Fujiwara documented
early neutrophilic infiltrate in all layers of the heart, including the valves. Inflammation
begins in the microvasculature (i.e., arterioles, capillaries, vasa vasorum, and venules)
and subsequently spreads to larger vessels, especially the coronary arteries.
150
In these lesions, infiltrating cells are mostly macrophages and IgA-secreting plasma
cells,
42
findings that may be unique to KD.
151
Endothelial cells express a variety of markers of activation, presumably as a result
of the high levels of proinflammatory cytokines that characterize the acute phase
of disease.
152
Some children have a lymphocytic myocarditis, with endomyocardial biopsy demonstrating
cellular infiltrates or myofibrosis that may persist for years in untreated cases.
153
Evolution of the cardiac lesions was detailed in the study by Fujiwara and Hamashima.
150
Pericarditis, myocarditis, and endocarditis were universal findings early in the disease,
but diminished as fibrosis of the myocardium became the predominant lesion in children
whose death occurred 40 days or more after onset. Coronary artery vasculitis predominated
early in the disease but was absent in those who died after 28 days of illness. Aneurysms,
thrombosis, and stenosis did not appear until 12 days of disease or later.
In a study of 262 children, Suzuki and colleagues
154
documented an equal frequency of aneurysms in the right and left coronary arteries,
but a higher propensity for development of segmental stenosis and occlusions in the
right coronary artery.
Using light and electron microscopy, Orenstein et al. reviewed autopsy and cardiac
transplant tissues from KD patients, and described three phases to the arteriopathy
of KD that differ in some ways from prior descriptions.
155
The first phase is characterized by a neutrophilic necrotizing arteritis that begins
in the endothelium and can cause saccular aneurysms as the process moves through the
walls of the arteries to the adventitia in the first 2 weeks of illness. This is followed
by a subacute or chronic vasculitis driven by lymphocytes, plasma cells, and eosinophils
that may last weeks to years and results in fusiform aneurysms. During the subacute
or chronic vasculitis, smooth muscle cells may be converted to myofibroblasts that
cause progressive stenosing lesions, leading to thrombosis.
155
Laboratory Examination
There are no specific diagnostic tests for KD, but at onset, evidence of inflammation
is manifested by elevation of C-reactive protein (CRP) and ESR, leukocytosis, and
a left shift in the white blood cell (WBC) differential count. Toxic granulation of
neutrophils is more frequent in children with KD than in those with other febrile
illnesses.
156
Occasionally, significant neutropenia occurs early
157
; this may be a marker for particularly severe disease. Thrombocytopenia and anemia
may herald the onset of macrophage activation syndrome (see Chapter 49).
158
Although platelet counts may be normal at the onset of disease, by the second week
of illness they characteristically rise and may reach 1,000,000/mm3 (reactive thrombocytosis)
in the most severe cases. Children with KD often present with a normocytic, normochromic
anemia; hemoglobin concentrations greater than 2 standard deviations below the mean
for age are found in half of patients within the first 2 weeks of illness.
11
Sterile pyuria is of urethral origin and therefore is missed on urinalyses obtained
by bladder aspiration or catheterization. The WBCs are mononuclear and are not detected
by dipstick tests for leukocyte esterase. Measurement of liver enzymes often reveals
elevated transaminase levels or mild hyperbilirubinemia due to intrahepatic congestion.
A few children develop obstructive jaundice from hydrops of the gallbladder or hepatic
vasculitis.
Cerebrospinal fluid (CSF) analysis typically displays a mononuclear pleocytosis with
normal glucose and protein. In a chart review of 46 children with KD, 39% were documented
to have elevated CSF WBC counts.
131
The median count was 22.5 cells/mm3 with 6% neutrophils and 91.5% mononuclear cells,
although cell counts as high as 320/mm3 with up to 79% neutrophils were reported.
Arthrocentesis of involved joints typically demonstrates synovial fluid WBC counts
of 50 to 300,000 WBC/mm3 consisting primarily of neutrophils.
Children with KD develop significant perturbations in serum lipid profiles beginning
during the subacute phase of illness. These abnormalities include elevated concentrations
of triglycerides and low-density lipoproteins, and depressed levels of high-density
lipoproteins.
159
They are most likely caused by widespread endothelial injury, and persistent abnormalities
in lipid profiles are more likely in those children with coronary artery abnormalities.
Ou et al. found that 1 year after the onset of KD, children with coronary artery aneurysms
were more likely to have depressed high-density lipoprotein cholesterol levels and
elevated high-sensitivity CRP levels than those KD patients who had normal coronary
arteries.
160
As with other sequelae of KD, normalization may take years in untreated children but
typically occurs within weeks or months after IVIG therapy.
ANCAs
161
and antibodies to endothelial cells
162
may be present late but not early in the disease.
67
Consequently, they have unclear pathological significance and are of little diagnostic
value. Other autoantibodies are usually absent. Elevated levels of von Willebrand
factor antigen indicate the presence of damaged endothelium.
163
Activation products of C3 and C4 have been demonstrated on erythrocytes (C3g) and
in the plasma (C4d),
164
suggesting the participation of complement in at least some of the manifestations
of the disease.
Treatment
General Approach
The child with suspected or definite KD should be admitted to the hospital for observation,
monitoring of cardiac status, and management of systemic manifestations (Box 35-3
). Initial evaluation of the heart should include an electrocardiogram to identify
dysrhythmias, signs of ischemia, or myocarditis. A baseline echocardiogram should
be performed to detect coronary artery vasculitis, ectasia, or aneurysms and to document
biventricular function. If the diagnosis is relatively certain (even if diagnostic
criteria are not met), and other diagnoses have been considered and excluded, treatment
should be initiated with aspirin and IVIG without further delay.
Box 35-3
Initial Evaluation and Management of Kawasaki Disease
Evaluation
•
General physical exam
•
Cardiac status (ECHO, ECG)
•
CNS status
•
Hematological and inflammatory parameters (CBC, differential, platelet count, ESR,
CRP)
•
Fluid and electrolyte status (AST, ALT, bilirubin, electrolytes, BUN, creatinine);
urinalysis
•
Ophthalmological status
•
Monitor cardiac status
•
Monitor CRP (ESR) and platelet count at 2-week intervals until stable, then 1-month
intervals until normal
•
Repeat echocardiogram at 6 to 8 weeks
Treatment
•
Aspirin:
•
If patient is febrile: 80 to 100 mg/kg/day in four doses
•
If patient is afebrile: 3 to 5 mg/kg/day in one dose
•
IVIG: 2 g/kg administered over 8 to 12 hours with premedications
•
Keep in hospital until afebrile for 24 hours or if there are complications
•
If fever persists, repeat IVIG once
•
If inadequate clinical response, consider corticosteroids (2 mg/kg/day, or 30 mg/kg/dose,
or infliximab 5 mg/kg [see discussion in this chapter])
•
Maintain low-dose aspirin until ESR and platelet count are normal if there have been
no coronary artery abnormalities; for 2 years if coronary abnormalities have resolved;
“forever” if coronary artery disease persists
ALT, Alanine transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen;
CNS, central nervous system; CRP, C-reactive protein; ECG, electrocardiogram; ECHO,
echocardiogram; ESR, erythrocyte sedimentation rate; IVIG, intravenous immunoglobulin.
Goals of Therapy
In addition to control of the acute inflammation and its symptoms, the goal of therapy
is to prevent long-term sequelae and, most importantly, coronary artery abnormalities.
The consequences of failure to appropriately treat a child with KD are so important
that, within reason and after very careful evaluation, error on the side of premature
or unnecessary therapy is preferable to delayed or missed therapy for a child for
whom the diagnosis is uncertain. The American Academy of Pediatrics and the AHA recommend
that children with KD should be treated with aspirin and IVIG during the first 10
days of the illness.7, 165
Treatment strategies also depend on the presence of coronary artery dilation, given
the long-term morbidity associated with this complication. Approximately half of
coronary artery aneurysms demonstrated by echocardiogram regress to normal lumen diameter
via myointimal proliferation in 1 to 2 years after illness onset, usually in aneurysms
smaller than 6 mm in diameter.
166
However, persistent vasodilatory abnormalities have been observed in arteries where
aneurysms resolved.
167
Giant coronary artery aneurysms, with an internal diameter larger than 8 mm, are associated
with the highest risk of morbidity and mortality. Up to one third of such aneurysms
become obstructed, leading to myocardial infarction, dysrhythmias, or sudden death.
168
Treatment with IVIG decreases the incidence of giant aneurysms by more than 98% and
the overall incidence of aneurysms by 85%.18, 169
Acute phase reactants and platelet counts do not return to normal for up to 2 months
after apparently successful treatment with IVIG, suggesting that vasculitis and endothelial
inflammation may not fully resolve, even when fever is controlled. IVIG-resistant
KD requires additional therapy, and questions remain whether initial treatment should
be more robust than IVIG alone, at least for some children at high risk of responding
incompletely to IVIG, aiming for anatomically and functionally normal vessels in everyone.
Aspirin
Aspirin was the first medication to be used for treatment of KD because of its antiinflammatory
and antithrombotic effects.
170
Antiinflammatory regimens using high-dose (>80 mg/kg/day)
7
or lower-dose (30 to 50 mg/kg/day)
170
aspirin have been recommended during the acute phase of the illness. After the fever
resolves, the dose is usually reduced to an antiplatelet range of 3 to 5 mg/kg/day.
These doses, well below the antiinflammatory level, have the effect of inhibiting
platelet adhesion to endothelium by curtailing platelet release of thromboxane A2
without suppressing prostacyclin production by endothelial cells.
171
This effect is thought to be beneficial in preventing thrombosis when platelet counts
are elevated, although no studies have demonstrated such a benefit clinically. In
the event of aspirin sensitivity, another antiplatelet agent, such as dipyridamole,
should be considered in patients at particular risk of developing thromboses. Unless
coronary artery abnormalities are detected by echocardiogram, aspirin is discontinued
after results of laboratory studies return to normal, usually within 2 months of disease
onset.
A meta-analysis found that high-dose and lower-dose aspirin regimens were associated
with a similar incidence of coronary artery abnormalities at 30 and 60 days after
disease onset.
172
Lee et al. enrolled 51 children with KD and treated them with standard doses of IVIG
but without concomitant use of acetylsalicylic acid (ASA) in the acute phase, and
compared them to a historical control group treated with IVIG plus high-dose ASA.
The ASA-treated group had shorter duration of fever as compared to the no-ASA group,
but there was no difference in IVIG resistance (17.1% vs. 15.7%, P = 1.000) or the
development of coronary artery lesions (7.8% vs. 3.9%, P = 0.514).
173
A retrospective study by Hsieh et al. had similar findings, although the duration
of fever was not different in the no-ASA group.
174
Although the necessity of using high-dose aspirin might be questioned because of the
rapid response to IVIG, all of the trials showing the benefit of IVIG were conducted
with children who also were receiving antiinflammatory doses of aspirin. There have
been no published comparisons of aspirin with other antiinflammatory agents, and it
is unclear whether salicylates are uniquely efficacious for this condition. For other
complications, such as treatment of prolonged arthritis, alternative antiinflammatory
agents may be used. The AHA warns against prescribing ibuprofen in children, as they
require protection from thrombosis because ibuprofen antagonizes the antiplatelet
effects of low-dose aspirin.7, 175
The risks of aspirin appear to be similar to those reported in other settings: chemical
hepatitis, transient hearing loss, and, rarely, Reye syndrome.
176
These risks may be increased in KD. Aspirin-binding studies have suggested that the
hypoalbuminemia of children with KD predisposes them to toxic levels of free salicylate,
despite measured (bound) values within the therapeutic range.
177
Intravenous Immunoglobulin
Furusho and co-workers
178
first reported that high-dose IVIG appeared to decrease the incidence of coronary
artery abnormalities. Newburger and colleagues
18
verified these findings in a 19-month, randomized, controlled clinical trial in 168
children with KD. Half of the children received IVIG (400 mg/kg/day on 4 consecutive
days) plus high-dose aspirin (100 mg/kg/day), and half of the children received aspirin
alone. IVIG reduced the incidence of coronary artery abnormalities by 78%, and no
child suffered serious adverse effects from the therapy, thereby confirming the remarkable
therapeutic potential of IVIG.
The initial IVIG treatment regimen was based on then-current protocols for treating
immune thrombocytopenic purpura. The question of whether this protocol was optimal
for KD was addressed in 1991.
179
Children were randomized to receive the traditional four-dose regimen or a single
dose of 2 g/kg of IVIG infused over 8 to 12 hours. Children receiving the larger,
single dose fared better. Meta-analyses have documented a dose-response benefit of
IVIG therapy in the range of 200 mg/kg to 2 g/kg.
180
IVIG is most effective in reducing the risk of coronary artery disease when administered
within 10 days of the onset of fever. Unfortunately, the diagnosis may remain in doubt
as this deadline approaches. In ambiguous cases, the physician may be guided by the
epidemiology of the disease. More than 50% of infants with KD present atypically (i.e.,
do not fulfill diagnostic criteria), and they have a very high incidence of aneurysms.
Thus, empiric treatment in very young children warrants serious consideration.
6
The mechanism of action of IVIG is uncertain, with studies adding induction of neutrophil
apoptosis
181
and reversal of inhibited lymphocyte apoptosis
182
to a long list of immunomodulatory effects of IVIG (Box 35-4
). The response is generally prompt, and temperature returns to normal in many children
even before the end of the IVIG infusion, with rapid clearing of the rash, mucositis,
and conjunctivitis. Irritability and emotional lability, however, may persist for
up to several weeks before resolving.
Box 35-4
Potential Effects of Intravenous Immunoglobulin in Kawasaki Disease
Specific Effects
•
Provides antibodies against infectious agent
•
Provides antibodies against circulating toxin
•
Provides antiidiotypic antibodies
Nonspecific Effects
•
Blockades Fc receptors
•
Accelerates clearance of activated complement fragments
•
Alters solubility characteristics of circulating immune complexes
•
Decreases soluble adhesion molecules (e.g., E-selectin, ICAM-1)
•
Upregulates activity of natural killer cells
•
Reverses immunoregulatory abnormalities by increasing suppressor T cells and decreasing
helper T cells and circulating B cells
•
Downregulates transcription of cytokine genes
•
Neutralizes activity of proinflammatory cytokines
•
Causes feedback inhibition of autoantibody synthesis
•
Reverses inhibited lymphocyte apoptosis
•
Induces neutrophil apoptosis
The greatest long-term concern about IVIG use is potential transmission of blood-borne
pathogens. Technical deficiencies in production led to more than 100 cases of hepatitis
C in recipients of a single brand of IVIG in 1994, although none was a child with
KD.
183
No cases of IVIG-transmitted infections have been reported since the institution,
in 1995, of current purification and processing practices, and no cases of IVIG-transmitted
human immunodeficiency virus (HIV) have ever been reported. Overall, the cost-benefit
analysis documents that IVIG treatment of KD is one of the most cost-effective medical
therapies available, leading to impressive short- and long-term savings.
184
Infusion reactions (fever, rash, nausea, and hypotension) occasionally accompany
IVIG administration and are best managed by slowing the rate of infusion and administering
diphenhydramine. With no viable alternative therapies, aggressive premedication with
corticosteroids, or even use of a different brand of IVIG, is preferable to foregoing
immunoglobulin. Rarely, a child might develop congestive heart failure during or after
infusion of the IVIG because of the high solute load and subsequent increase in intravascular
volume. Slowing the infusion rate and administration of furosemide are usually the
only treatments required. Treatment with IVIG leads to improvement in myocardial contractility
and is almost invariably adequate therapy.
7
Hemolysis is uncommon, but occasionally it may be severe, requiring transfusion.
185
Headache up to 72 hours after the infusion is common, especially in older patients.
Such children may require low-dose opiates for relief.
186
Virtually all data concerning the role of IVIG are limited to treatment during the
first 10 days of illness. This is not to say that treatment after 10 days of illness
is ineffective or contraindicated; it is merely inadequately studied. In a report
of 16 children with coronary artery aneurysms treated a mean of 17 days after the
onset of fever, echocardiogram showed there was a trend toward resolution of abnormalities.
187
The American Academy of Pediatrics cautiously recommends IVIG for children beyond
the tenth day of illness with “manifestations of continuing inflammation,” and such
an approach appears prudent.
165
Questions have arisen concerning very early treatment of KD.
188
Tse and colleagues,
189
on the other hand, reported that IVIG given on or before the fifth day of illness
resulted in fewer coronary artery abnormalities at the 1-year follow-up assessment.
Thus, decisions about the optimal date for treating with IVIG are best made based
on a patient's clinical status and the certainty of the diagnosis of KD rather than
anticipated advantages of administration on a particular day of disease.
Prediction of IVIG Resistance
The clinical importance of predicting which children will suffer from cardiac sequelae
from KD has led to the creation of several risk scores for IVIG resistance. In a retrospective
series from Japan, Fukunishi and colleagues
190
found higher serum levels of CRP, lactate dehydrogenase, and bilirubin to be predictive
of failure to respond to IVIG. More recently, Kobayashi and colleagues reported on
several factors that were associated with decreased responsiveness to IVIG, and therefore
increased risk of coronary artery abnormalities: hyponatremia; elevated hepatic transaminase
and CRP; a high percentage of bands on the WBC count differential; a platelet count
of 300,000 or less; short duration between fever onset and diagnosis (4 days or less);
and being younger than 12 months of age at onset.
191
Egami et al.
192
and Sano et al.
193
have also constructed risk scores for IVIG resistance utilizing similar parameters.
Unfortunately, application of these risk scores did not accurately identify all children
at risk for IVIG resistance and coronary artery abnormalities in a North American
cohort.
194
In a Canadian study, Han and colleagues
188
could not identify any difference in laboratory parameters between responders and
nonresponders. Confirming the importance of controlling inflammation in KD, Mori and
co-workers
195
reported that a rise in the WBC count and CRP level after IVIG infusion are independent
predictors of coronary artery abnormalities.
Glucocorticoids
Glucocorticoids, the preferred initial treatment for other forms of vasculitis, were
considered unsafe in KD for many years following the early descriptions of the disease.
This is based primarily on a study
196
that demonstrated an extraordinarily high incidence of coronary artery aneurysms (11
of 17 patients) in a group that received oral prednisolone at a dose of 2 to 3 mg/kg/day
for at least 2 weeks, followed by 1.5 mg/kg/day for an additional 2 weeks. Interestingly,
seven patients in the same study received prednisolone plus aspirin, and none developed
aneurysms. In fact, no subsequent study has indicated that corticosteroids are harmful
when used either with IVIG or as an alternative to IVIG therapy. Corticosteroids in
KD have been studied both as primary therapy and “rescue” therapy, and doses have
ranged from pulse doses of 30 mg/kg (maximum of 1 g) to conventional antiinflammatory
doses (2 mg/kg/day).
Potential benefits of corticosteroids as rescue therapy in KD have been reported.
Initially, two retrospective analyzes supported the use of corticosteroids in children
who were unresponsive to two doses of IVIG or who relapsed after such therapy.197,
198 Hashino and colleagues
199
also found a beneficial effect of glucocorticoids in KD in a prospective trial. Children
who had failed to respond to two doses of IVIG were randomized to receive a third
dose of IVIG or pulse-dose methylprednisolone. Patients who received methylprednisolone
had a significantly shorter duration of fever, and although transient coronary artery
dilation was associated with glucocorticoid therapy, there was no overall difference
in the incidence of coronary artery abnormalities between groups. Recently, Kobayashi
et al.
200
retrospectively reviewed 359 consecutive KD patients over 12 years who failed to respond
to first-line therapy of IVIG. They compared outcomes of children who received a second
dose of IVIG versus a second dose of IVIG plus prednisolone versus prednisolone as
monotherapy (maximum dose of 2 mg/kg/day for all children receiving steroids). They
found that outcomes were better in the IVIG + prednisolone group with decreased need
for subsequent treatments (aOR 0.16, 95% confidence interval [CI] 0.09-0.31), and
fewer coronary artery abnormalities at 1 month (aOR 0.40, 95% CI 0.18-0.91) than the
IVIG group. However, the treatment regimens were selected arbitrarily in this retrospective
study. A prospective study is likely needed to assess the role of corticosteroids
as rescue therapy.
Might steroids be more effective if administered earlier in the course of KD? Shinohara
and colleagues
201
retrospectively reviewed the results in almost 300 patients with acute KD seen between
1982 and 1998 who were treated before the tenth day of illness. All patients received
aspirin, dipyridamole, and propranolol. The addition of prednisolone therapy, either
alone or with IVIG, was associated with a significantly shorter duration of fever
and a lower prevalence of coronary artery aneurysms. No adverse reactions were recorded
for any therapy. A prospective study suggested benefit as well: Inoue
202
reported that the frequency of coronary artery abnormalities in children treated with
IVIG plus prednisolone at a dose of 2 mg/kg/d was lower than in those treated with
IVIG alone. Three other studies197, 203, 204 have shown that children treated with
intravenous methylprednisolone (IVMP) (or dexamethasone) plus IVIG had a faster resolution
of fever, more rapid improvement in the markers of inflammation, and a shorter length
of hospitalization than those who received IVIG alone. Two of these studies had insufficient
statistical power to detect a potential benefit of glucocorticoid therapy on coronary
artery outcomes. The third trial, by Newburger and colleagues, found no significant
difference in the frequency or severity of coronary artery lesions between treatment
groups at the 1- or 5-week follow-up. Interestingly, however, post hoc analysis suggested
that children who ultimately failed to respond to an initial dose of IVIG were less
likely to develop coronary artery aneurysms if their initial therapy had included
IVMP.
Following up on this finding, the Osaka Kawasaki Disease Study Group
205
conducted a comparative trial of IVIG versus IVIG + IVMP in children with KD who were
regarded as being at high risk to be nonresponse to IVIG.
193
Patients were given heparin (10 U/kg/hour) for 48 hours beginning 2 hours before receiving
IVMP (30 mg/kg), followed by IVIG (2 g/kg). Aspirin (30 mg/kg/d) was started at the
end of the heparin infusion and reduced to 10 mg/kg/day after resolution of fever.
Therapy was effective in 44% of those given IVIG alone compared with 66% of those
receiving both IVIG and IVMP. Coronary artery abnormalities, including aneurysms,
were significantly less frequent in the IVIG + IVMP group (24%) compared with the
IVIG-alone group (46%).
In a meta-analysis of eight studies, Wooditch and Aronson concluded that the incidence
of coronary artery aneurysms was reduced by the addition of corticosteroids to therapeutic
regimens that included aspirin.
198
However, a subsequent meta-analysis of four studies that evaluated primary treatment
of KD with corticosteroids found that IVIG resistance was less common in those treated
with steroids as primary therapy (OR 0.48, 95% CI 0.24-0.95), but coronary outcomes
did not differ.
206
The most definitive trial to date regarding corticosteroids in combined primary therapy
with IVIG was the RAISE trial by Kobayashi et al. in 2012.
207
There were 248 patients were enrolled in this multicenter, prospective, randomized,
open label, blinded end points trial. All patients enrolled had a Kobayashi score
or 5 or greater,
191
and therefore were considered to be at high risk for IVIG resistance. Of note, patients
on day 9 or later of illness were excluded, as were patients with coronary artery
abnormalities on baseline echocardiogram. Patients were randomized to standard therapy
with IVIG and ASA versus IVIG plus prednisolone at a dose of 2 mg/kg/day. The corticosteroid
was initially given intravenously for 5 days, which was changed to oral dosing if
the patient's fever abated, and then tapered following normalization of the CRP. The
primary end point of the trial was defined as coronary artery abnormalities per JPH
criteria seen on two-dimensional (2D) echocardiography in the steroid versus IVIG
alone groups at weeks 1, 2, or 4. A significant difference in coronary artery abnormalities
between the groups at the interim analysis, favoring administration of steroids with
IVIG (3% [n = 4] vs. 23% [n = 28], P < 0.0001), led to early termination of the study.
Secondary end points included incidence of coronary artery abnormalities at week 4,
z scores of coronary arteries, incidence of need for rescue therapy, duration of fever
after enrollment, and serum CRP concentrations at weeks 1 and 2. All secondary end
points were also met, a remarkable achievement. Of note, although the overall incidence
of coronary artery abnormalities in the IVIG group was high at 23% during the study
period, as would be expected in this group of high-risk patients, the maximum z scores
were relatively low, between 2.26 and 2.32.
207
Challenges in determining the optimal use of corticosteroid treatment in KD remain.
An accurate, easily applicable risk score has not been constructed to effectively
stratify children with KD in North America and Europe who are at increased risk of
developing coronary artery abnormalities. Furthermore, it remains unclear whether
corticosteroids are best used as intensification of primary therapy for all KD patients
at a time when the vascular walls of the arteries may be particularly vulnerable,
or as rescue therapy for children who fail conventional therapy and are at higher
risk for coronary artery abnormalities.
Anti-TNF Agents
Levels of TNF-α are markedly increased in children with KD, especially in those who
develop coronary artery lesions.208, 209 As such, infliximab, a monoclonal antibody
to TNF-α, has been the subject of trials in children with KD, both as rescue therapy
as well as primary therapy.
A prospective randomized multicenter comparison of the effectiveness of IVIG (2 g/kg)
and infliximab (5 mg/kg) in children who had not responded to an initial infusion
of IVIG
210
showed that both agents were equally safe and well tolerated. Hirono and colleagues
211
also found that infliximab was effective in controlling fever but did not completely
prevent coronary artery changes, although single case reports document resolution
of aneurysms following infliximab therapy in some patients.212, 213 A retrospective
two-center comparison of KD patients resistant to initial therapy with IVIG who were
treated with either methylprednisolone (30 mg/kg) or infliximab (5 mg/kg) found that
infliximab-treated patients had less fever and fewer days in the hospital, but there
were no differences in coronary artery outcomes between the treatment groups.
214
Recently, Tremoulet et al. explored the utility of administering infliximab (5 mg/kg)
as primary therapy with IVIG.
215
There were 196 patients enrolled in a phase 3, randomized, double-blind, placebo-controlled
trial at two centers. The primary end point of a difference in IVIG resistance between
patients receiving combined therapy with IVIG and infliximab, and those receiving
IVIG alone, was not met (11.2% vs. 11.3%, P = 0.81). Patients treated with infliximab
had fewer days of fever and reduced inflammatory markers. The z score of the left
anterior descending artery was significantly decreased in the infliximab group as
compared with the placebo group at week 2 (P = 0.45). However, coronary outcomes at
week 5 did not differ between treatment groups. There were no serious adverse events
attributed to infliximab during the trial. At this time, the use of infliximab in
the treatment of patients with KD remains essentially center-dependent, though convincing
evidence of a beneficial effect on coronary artery outcomes is lacking.
Other Therapeutic Approaches
Therapies that are effective in other forms of vasculitis have been used in KD. Pentoxifylline
was alleged to be effective in preventing coronary artery aneurysms,
216
but demonstration of flaws in the analysis of the data in this study
217
led to the conclusion that it is ineffective. Similarly, the human trypsin inhibitor,
Ulinastatin, has been the subject of studies from Japan. Its efficacy in preventing
coronary artery disease in KD is not convincing.
218
The recent data regarding the potential role of T cells in KD59, 72 have led researchers
to prescribe cyclosporine, a potent suppressor of T-cell activity through the nuclear
factor of activated T-cells (NFAT) pathway. Suzuki et al.
219
studied 28 patients treated with cyclosporine A (CyA, 4 to 8 mg/kg/day) for refractory
KD, defined as persistent fever after two doses of IVIG. The fevers of 18 of 28 patients
subsided within 3 days of starting CyA. Four patients developed aneurysms, one of
which was a giant aneurysm. Hyperkalemia occurred in nine patients, but no serious
adverse events were reported. Tremoulet et al. also evaluated the use of calcineurin
inhibitors in IVIG-resistant KD.
220
All 10 patients had already received rescue therapy in the form of an additional dose
of IVIG (10 patients), pulsed methylprednisolone (3 patients), and infliximab (4 patients).
Following treatment with a calcineurin inhibitor, all 10 patients reported the subsidence
of fever. Seven of the patients experienced rapid resolution of the fever within 24
hours of starting treatment. Four of the patients had developed coronary artery aneurysms
prior to therapy with a calcineurin inhibitor; all improved thereafter.
The use of statins has been explored in patients with significant cardiovascular sequelae
from KD, given their potential beneficial effects on vascular reactivity and remodeling
as well as their antiinflammatory effects.221, 222 A very small study of 11 KD patients
with coronary artery aneurysms treated with simvastatin for 3 months reported a significant
reduction in the high-sensitivity (hs)-CRP level and improvement in flow-mediated
dilation.
223
Niedra et al. evaluated the safety of atorvastatin by following 20 patients with coronary
artery aneurysms for a median of 2.5 years while treated with atorvastatin (5 to 10 mg
daily).
224
Almost half of the patients had at least one episode of hypocholesterolemia, and two
required a lowered dose. Mild transaminitis occurred in seven of the patients; only
one patient had increased creatine phosphokinase level. They concluded that use of
atorvastatin was safe with close monitoring.
224
The potential role for cyclophosphamide
225
in KD is extremely limited, but it may be useful in cases with persistent active disease
that is unresponsive to conventional therapy. In fact, children with prolonged inflammation
ascribed to KD may be similar to children with polyarteritis nodosa, in which longer-term
immunosuppression with cyclophosphamide is standard therapy.
226
A dramatic response to plasmapheresis in refractory cases of KD also has been reported,
227
but the technical limitations and potential hazards of this therapy are considerable.
It should be reserved for children with active inflammation who have failed all available
medical interventions, including multiple doses of IVIG, intravenous methylprednisolone,
and TNF inhibition. There have been conflicting reports of the efficacy of abciximab,
a monoclonal antibody that inhibits platelet glycoprotein IIb/IIIa receptor. In one
study,
228
there was an increased resolution of aneurysms in patients with KD who received abciximab
compared with those who received conventional treatment. However, a second study
229
could not duplicate these findings.
Treatment of Relapses
Fever returns within 48 hours of treatment with IVIG in 10% to 20% of children, indicating
failure to suppress the underlying inflammatory process. Because prolonged fever is
an independent risk factor for the development of coronary artery aneurysms, current
treatment protocols generally recommend retreatment with a second dose of IVIG (2 g/kg).
6
Those who fail to respond to a second dose—up to one third of patients in some studies
230
—are at extremely high risk of developing coronary artery aneurysms.
199
As noted above, use of corticosteroids appears to have the most convincing evidence
of benefit in children resistant to IVIG, although definitive evidence for preference
of one regimen over another is lacking. Therapeutic strategies include intravenous
methylprednisolone (30 mg/kg/day for 1 to 3 days)
188
prednisolone at 2 mg/kg/day,
200
or infliximab (5 mg/kg).
231
Regardless of which approach is selected, treatment should continue until fever resolves
and the CRP is normal. Frequent monitoring of the coronary arteries should be pursued
until children have fully recovered.
Prevention and Management of Thromboses
The risk of thrombosis of coronary or other arteries depends on the degree of vascular
damage. In all patients with KD, irrespective of the demonstration of coronary artery
abnormalities, low-dose (3 to 5 mg/kg/day) aspirin should be continued until the ESR
and platelet counts have normalized. Children with coronary artery abnormalities demonstrated
by echocardiography are often treated with antithrombotic agents, such as low-dose
aspirin, for as long as the abnormalities persist (Table 35-2
). Children with large aneurysms are given warfarin or low-molecular-weight heparin
to induce anticoagulation.
TABLE 35-2
Recommendations for Long-Term Follow-Up
RISK LEVEL
PHARMACOLOGICAL THERAPY
PHYSICAL ACTIVITY
FOLLOW-UP AND DIAGNOSTIC TESTING
INVASIVE TESTING
I (no coronary artery changes at any stage of illness)
None beyond first 6-8 weeks
No restrictions beyond first 6-8 weeks
Cardiovascular risk assessment counseling at 5-year intervals
None recommended
II (transient coronary artery ectasia disappears within first 6-8 weeks)
None beyond first 6-8 weeks
No restrictions beyond first 6-8 weeks
Cardiovascular risk assessment counseling at 3- to 5-year intervals
None recommended
III (1 small to medium coronary artery aneurysm/major coronary artery)
Low-dose aspirin (3-5 mg/kg aspirin/day), at least until aneurysm regression documented
For patients <11 years old, no restriction beyond first 6-8 weeks; patients 11-20
years old, physical activity guided by biennial stress test, evaluation of myocardial
perfusion scan; contact or high-impact sports discouraged for patients taking antiplatelet
agents
Annual cardiology follow-up with echocardiogram + electrocardiogram, combined with
cardiovascular risk assessment, counseling; biennial stress test/evaluation of myocardial
perfusion scan
Angiography, if noninvasive test suggests ischemia
IV (≥1 large or giant coronary artery aneurysm, or multiple or complex aneurysms in
same coronary artery, without obstruction)
Long-term antiplatelet therapy and warfarin (target international normalized ratio
2.0-2.5) or low-molecular-weight heparin (target: antifactor Xa level 0.5-1.0 U/mL)
should be combined in giant aneurysms
Contact or high-impact sports should be avoided because of risk of bleeding; other
physical activity recommendations guided by stress test/evaluation of myocardial perfusion
scan outcome
Biannual follow-up with echocardiogram + electrocardiogram; annual stress test/evaluation
of myocardial perfusion scan
First angiography at 6-12 months or sooner if clinically indicated; repeated angiography
if noninvasive test, clinical, or laboratory findings suggest ischemia; elective repeat
angiography under some circumstances
V (coronary artery obstruction)
Long-term low-dose aspirin; warfarin or low-molecular-weight heparin if giant aneurysm
persists; consider use of β-blockers to reduce myocardial O2 consumption
Contact or high-impact sports should be avoided because of risk of bleeding; other
physical activity recommendations guided by stress test/myocardial perfusion scan
outcome
Biannual follow-up with echocardiogram and electrocardiogram; annual stress test/evaluation
of myocardial perfusion scan
Angiography recommended to address therapeutic options
From Newburger, Takahashi, Gerber, et al., Diagnosis, treatment and long-term management
of Kawasaki disease: a statement for health professionals from the committee on rheumatic
fever, endocarditis and Kawasaki disease. Council on Cardiovascular Disease in the
Young: American Heart Association, Pediatrics 114 (2004) 1708–1733.
When injured coronary arteries become obstructed (risk level V), in addition to anticoagulation,
various therapies have been attempted to restore circulation. Should the obstruction
occur within 6 weeks of the onset of illness, control of vascular inflammation with
IVIG and other agents is an essential prerequisite to arterial reperfusion. Thereafter,
treatments may include thrombolytic therapy for arterial thrombosis or vasodilators
if tissue viability is primarily threatened by vasospasm. Urokinase, streptokinase,
and tissue-type plasminogen have all been used for the lysis of coronary artery thromboses.
Similarly, peripheral arterial obstruction may be corrected by thrombolysis, after
which perfusion is maintained with heparin followed by a chronic oral anticoagulant
regimen. If these treatments fail, a variety of invasive approaches have been suggested,
including percutaneous transluminal coronary angioplasty
232
and coronary artery bypass grafting.
233
A small number of children with particularly severe coronary artery disease due to
KD have required cardiac transplantation.
234
Monitoring Cardiac Status
There is no universal agreement about the timing and frequency of echocardiographic
monitoring of patients with KD. Most protocols take into account the development of
coronary artery aneurysms, which occur most frequently between the second and the
eighth weeks after the onset of fever. It is recommended that the initial echocardiogram
be obtained at the time a diagnosis of KD is suspected, and that each child with KD
have a repeat echocardiography at 2 weeks and 6 weeks following illness.
235
Patients should also have repeated clinical examinations during the first 2 months
to detect dysrhythmias, congestive heart failure, valvular insufficiency, or myocarditis.
236
Further follow-up is individualized, with more frequent studies performed in children
with demonstrated coronary artery abnormalities (see Table 35-2).
Children whose coronary arteries have always been normal (risk level I) or are normal
by echocardiographic criteria 1 to 2 months after the acute illness (risk level II)
are regarded as healthy, and no further intervention is recommended after the 8-week
follow-up assessment. In view of possible chronic abnormalities in endothelial function,
however, many physicians consider a history of KD to be a risk factor for the development
of coronary artery disease later in life.
237
They counsel modification of other atherosclerotic risk factors and continue to monitor
children once every 5 years.
Single small- to medium-sized aneurysms (risk level III) usually resolve as determined
by echocardiographic criteria, although this is not always the case. Healing occurs
by fibrointimal proliferation, often accompanied by calcification, and vascular reactivity
does not return to normal despite a grossly normal appearance.
238
This point is highlighted by a report of the sudden death of a -year-old child 3 months
after the child's dilated coronary arteries had regained a normal echocardiographic
appearance.
239
Autopsy revealed obliteration of the lumen of the left anterior descending coronary
artery due to fibrosis, with evidence of ongoing active inflammation in the epicardial
arteries. Such reports emphasize the need for confirmation of complete response to
therapy in children who have had KD.
Giant aneurysms with an internal diameter of at least 8 mm represent a significant
risk for morbidity and mortality, including a 35% chance of infarction (risk level
IV).
168
These children are followed more closely and are treated with more aggressive antithrombotic
and anticoagulation regimens.
Disease Course and Prognosis
Although standard therapy with IVIG and aspirin given within the first 10 days of
illness greatly improves outcomes, approximately 5% of children still develop coronary
artery aneurysms, and more children demonstrate coronary artery ectasia.
7
The mortality rate has dropped steadily as the diagnosis and treatment have improved.
Currently, the rate is about 0.1% in the United States and Japan.239, 240 Recurrent
disease after full recovery from a first episode of KD is rare, but it does occur.
In Japan, the recurrence rate is 3.6%,
31
with a higher incidence of cardiac complications during the second episode.
241
In the United States, the rate of recurrence is lower.
There have been two recent studies from Japan of long-term outcomes in KD cases complicated
by giant coronary artery aneurysms. Suda et al. reviewed the case records of 76 patients
with giant coronary artery aneurysms and found that the 30-year survival rate was
88%. However, there was a nearly 60% cumulative coronary intervention rate at 25 years
from onset, indicating that these patients carry significant morbidity in terms of
multiple procedures.
242
Tsuda reported similar survival rates in patients with giant coronary artery aneurysms
followed for up to 3 decades, and noted that the long-term outcomes were worse for
those patients with involvement of both the right coronary and the left coronary arteries.
243
As mentioned previously, whether children with normal coronary artery dimensions throughout
their illness are at higher risk for atherosclerotic disease later in life remains
an area of ongoing research. Studies to date have been conflicting.244, 245, 246,
247, 248 However, when standardized mortality ratios were calculated in 2009 for individuals
in Japan who were diagnosed with KD during the years 1982–1992 and who had no cardiac
sequelae, the mortality ratios of the KD patients showed no increases as compared
to the general population.
249
Definitive data regarding long-term outcomes in KD patients who always have normal
coronary arteries will likely be established as the KD cohorts in Japan reach middle
age.
Related collections
Most cited references192
- Record: found
- Abstract: found
- Article: not found
Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Statement for Health Professionals From the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association
Jane W Newburger, Masato Takahashi, Michael Gerber … (2004)
- Record: found
- Abstract: found
- Article: not found
Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association.
J Newburger, M. Takahashi, M Gerber … (2004)
- Record: found
- Abstract: found
- Article: not found
Prediction of intravenous immunoglobulin unresponsiveness in patients with Kawasaki disease.
Tohru Kobayashi, Yoshinari Inoue, Kazuo Takeuchi … (2006)