- Record: found
- Abstract: found
- Article: not found
Structures of Hepatitis B Virus Cores Presenting a Model Epitope and Their Complexes with Antibodies

Read this article at

Abstract
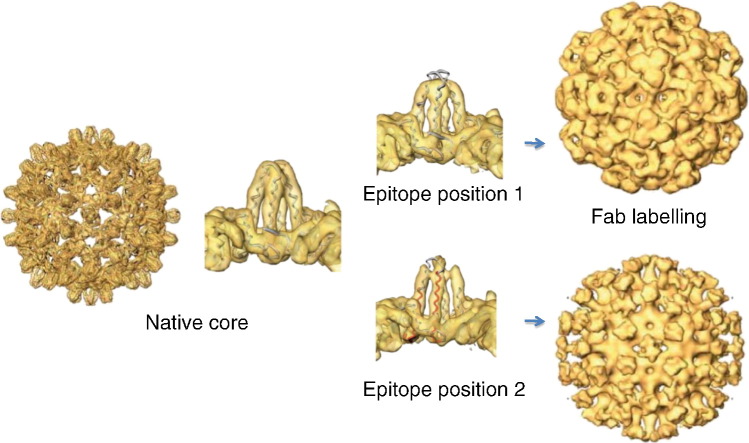
The core shell of hepatitis B virus is a potent immune stimulator, giving a strong neutralizing immune response to foreign epitopes inserted at the immunodominant region, located at the tips of spikes on the exterior of the shell. Here, we analyze structures of core shells with a model epitope inserted at two alternative positions in the immunodominant region. Recombinantly expressed core protein assembles into T = 3 and T = 4 icosahedral shells, and atomic coordinates are available for the T = 4 shell. Since the modified protein assembles predominantly into T = 3 shells, a quasi-atomic model of the native T = 3 shell was made. The spikes in this T = 3 structure resemble those in T = 4 shells crystallized from expressed protein. However, the spikes in the modified shells exhibit an altered conformation, similar to the DNA containing shells in virions. Both constructs allow full access of antibodies to the foreign epitope, DPAFR from the preS1 region of hepatitis B virus surface antigen. However, one induces a 10-fold weaker immune response when injected into mice. In this construct, the epitope is less constrained by the flanking linker regions and is positioned so that the symmetry of the shell causes pairs of epitopes to come close enough to interfere with one another. In the other construct, the epitope mimics the native epitope conformation and position. The interaction of native core shells with an antibody specific to the immunodominant epitope is compared to the constructs with an antibody against the foreign epitope. Our findings have implications for the design of vaccines based on virus-like particles.
Abstract
Highlights
► The HBV core shell is highly immunogenic and is being used as a vaccine carrier. ► Insertion of model epitopes into the immunodominant region changes the structure. ► Alternative positions of an epitope give different structures and immunogenicity. ► The structural differences lead to different labeling with antibody fragments. ► We conclude that three‐dimensional structural analysis will be important in vaccine design.
Related collections
Most cited references44
- Record: found
- Abstract: found
- Article: not found
The crystal structure of the human hepatitis B virus capsid.
- Record: found
- Abstract: found
- Article: not found
Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy.
- Record: found
- Abstract: found
- Article: not found