- Record: found
- Abstract: found
- Article: found
Complement in Thrombotic Microangiopathies: Unraveling Ariadne's Thread Into the Labyrinth of Complement Therapeutics
Read this article at
Abstract
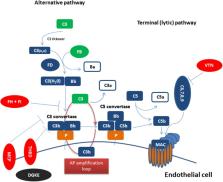
Thrombotic microangiopathies (TMAs) are a heterogeneous group of syndromes presenting with a distinct clinical triad: microangiopathic hemolytic anemia, thrombocytopenia, and organ damage. We currently recognize two major entities with distinct pathophysiology: thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS). Beyond them, differential diagnosis also includes TMAs associated with underlying conditions, such as drugs, malignancy, infections, scleroderma-associated renal crisis, systemic lupus erythematosus (SLE), malignant hypertension, transplantation, HELLP syndrome (hemolysis, elevated liver enzymes, and low platelets), and disseminated intravascular coagulation (DIC). Since clinical presentation alone is not sufficient to differentiate between these entities, robust pathophysiological features need to be used for early diagnosis and appropriate treatment. Over the last decades, our understanding of the complement system has evolved rapidly leading to the characterization of diseases which are fueled by complement dysregulation. Among TMAs, complement-mediated HUS (CM-HUS) has long served as a disease model, in which mutations of complement-related genes represent the first hit of the disease and complement inhibition is an effective and safe strategy. Based on this knowledge, clinical conditions resembling CM-HUS in terms of phenotype and genotype have been recognized. As a result, the role of complement in TMAs is rapidly expanding in recent years based on genetic and functional studies. Herein we provide an updated overview of key pathophysiological processes underpinning complement activation and dysregulation in TMAs. We also discuss emerging clinical challenges in streamlining diagnostic algorithms and stratifying TMA patients that could benefit more from complement modulation. With the advent of next-generation complement therapeutics and suitable disease models, these translational perspectives could guide a more comprehensive, disease- and target-tailored complement intervention in these disorders.
Related collections
Most cited references123
- Record: found
- Abstract: found
- Article: not found
Syndromes of thrombotic microangiopathy.
- Record: found
- Abstract: found
- Article: not found
Haemolytic uraemic syndrome
- Record: found
- Abstract: found
- Article: not found