- Record: found
- Abstract: found
- Article: found
Phenotypic Variability of c.436delC DCAF17 Gene Mutation in Woodhouse-Sakati Syndrome
Read this article at
Abstract
Case series
Patients: 38, female • 28, female • 41, female • 18, female • 23, male
Final Diagnosis: Woodhouse-Sakati syndrome
Symptoms: Hypogonadism • dystonia • alopecia • hearing loss • diabetes
Medication: —
Clinical Procedure: —
Specialty: Endocrinology and Metabolic
Background:
Woodhouse-Sakati syndrome (WSS) is a rare autosomal recessive genetic condition that was first described in 1983. Since its original description, approximately 50 cases have been reported with various clinical signs and symptoms. Characteristics include progressive neurologic deterioration with extrapyramidal involvement and polyendocrinopathy most notable for hypogonadism starting in early adolescence. Clinical presentation is variable, and a subset of patients may have additional features, such as premature aging, alopecia, distinctive facial features, cognitive impairment, or deafness.
Case Report:
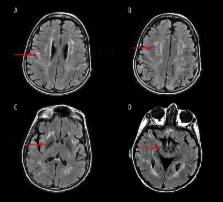
We illustrate the phenotypic variability of 5 patients with WSS due to the previously reported homozygous single nucleotide deletion c.436delC in the DCAF17 gene, identified in 2008. Despite identical genetic alteration, our 5 patients had various clinical features among them and compared with previously reported cases with the same pathogenic mutation.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome.
- Record: found
- Abstract: found
- Article: not found
A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities.
- Record: found
- Abstract: found
- Article: not found