- Record: found
- Abstract: found
- Article: found
The Remarkable Amphoteric Nature of Defective UiO‐66 in Catalytic Reactions
Read this article at
Abstract
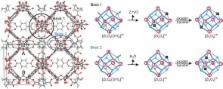
One of the major requirements in solid acid and base catalyzed reactions is that the reactants, intermediates or activated complexes cooperate with several functions of catalyst support. In this work the remarkable bifunctional behavior of the defective UiO‐66(Zr) metal organic framework is shown for acid‐base pair catalysis. The active site relies on the presence of coordinatively unsaturated zirconium sites, which may be tuned by removing framework linkers and by removal of water from the inorganic bricks using a dehydration treatment. To elucidate the amphoteric nature of defective UiO‐66, the Oppenauer oxidation of primary alcohols has been theoretically investigated using density functional theory (DFT) and the periodic approach. The presence of acid and basic centers within molecular distances is shown to be crucial for determining the catalytic activity of the material. Hydrated and dehydrated bricks have a distinct influence on the acidity and basicity of the active sites. In any case both functions need to cooperate in a concerted way to enable the chemical transformation. Experimental results on UiO‐66 materials of different defectivity support the theoretical observations made in this work.
Related collections
Most cited references38
- Record: found
- Abstract: not found
- Article: not found
Efficient iterative schemes forab initiototal-energy calculations using a plane-wave basis set
- Record: found
- Abstract: not found
- Article: not found
Generalized Gradient Approximation Made Simple.
- Record: found
- Abstract: found
- Article: not found